University of Bonn, Bonn-Aachen International Center for IT, Bonn, Germany.
PLoS One. 2011;6(10):e25364. doi: 10.1371/journal.pone.0025364. Epub 2011 Oct 25.
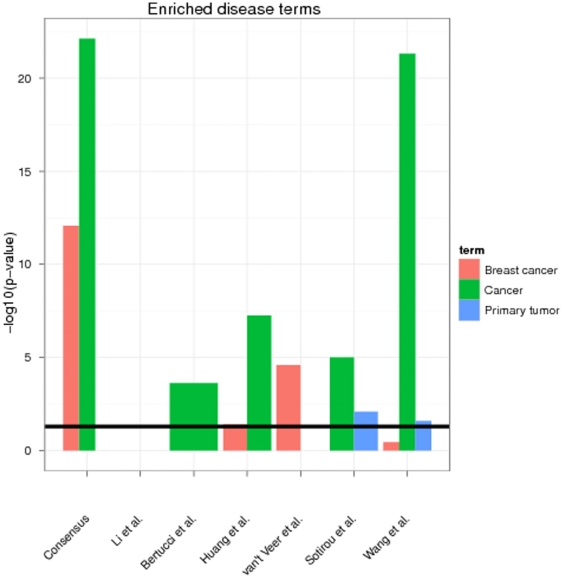
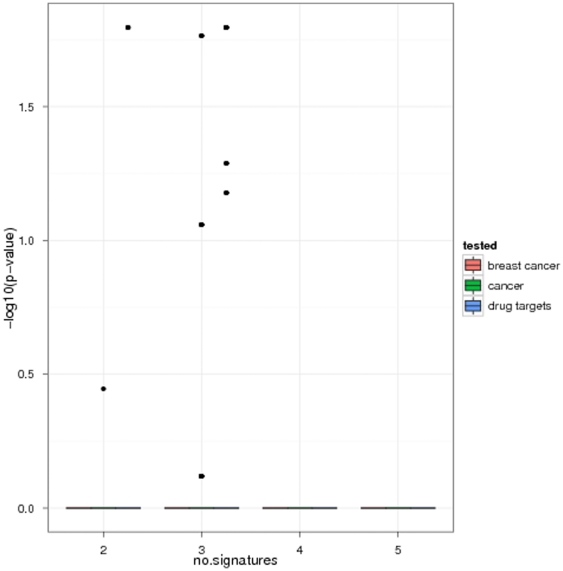
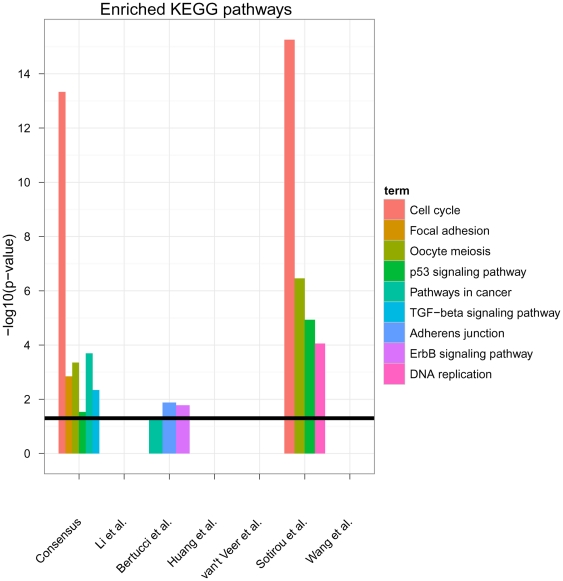
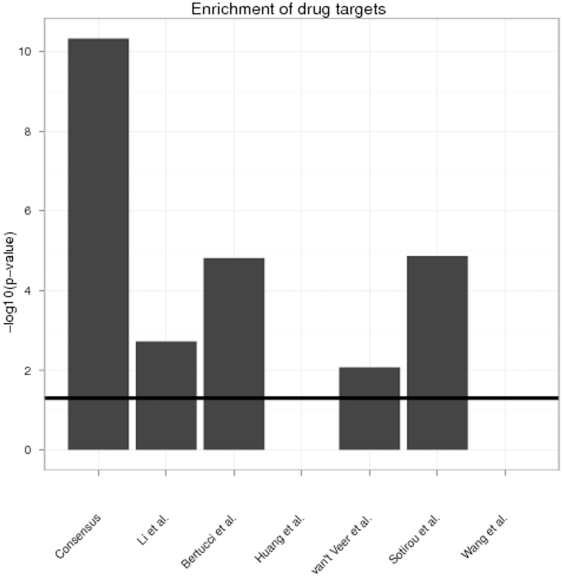
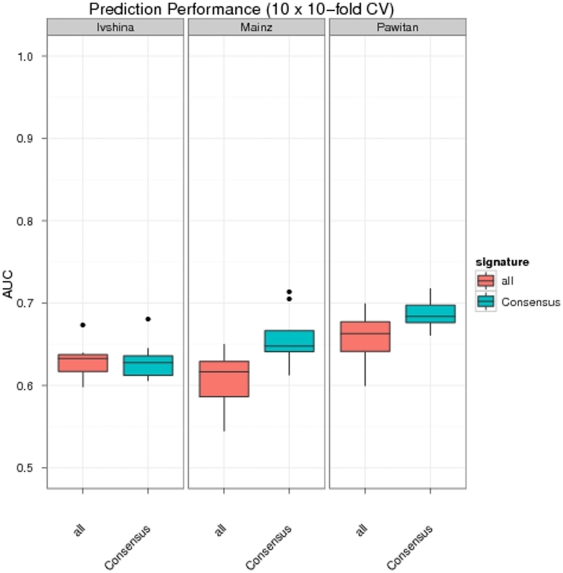
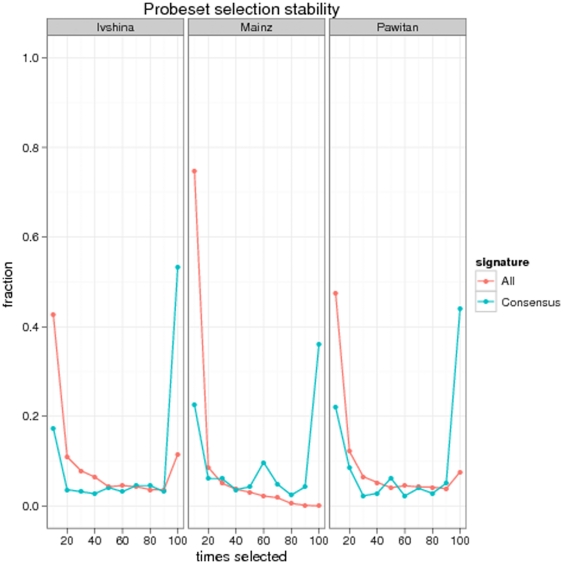
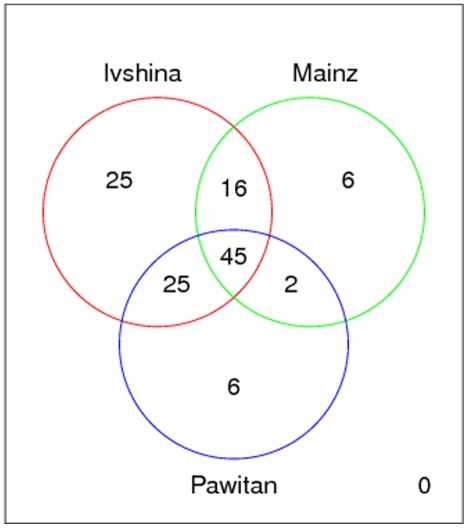
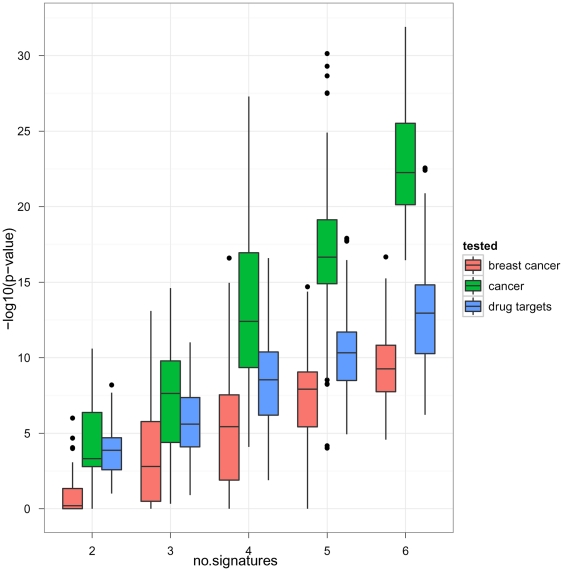
Diagnostic and prognostic biomarkers for cancer based on gene expression profiles are viewed as a major step towards a better personalized medicine. Many studies using various computational approaches have been published in this direction during the last decade. However, when comparing different gene signatures for related clinical questions often only a small overlap is observed. This can have various reasons, such as technical differences of platforms, differences in biological samples or their treatment in lab, or statistical reasons because of the high dimensionality of the data combined with small sample size, leading to unstable selection of genes. In conclusion retrieved gene signatures are often hard to interpret from a biological point of view. We here demonstrate that it is possible to construct a consensus signature from a set of seemingly different gene signatures by mapping them on a protein interaction network. Common upstream proteins of close gene products, which we identified via our developed algorithm, show a very clear and significant functional interpretation in terms of overrepresented KEGG pathways, disease associated genes and known drug targets. Moreover, we show that such a consensus signature can serve as prior knowledge for predictive biomarker discovery in breast cancer. Evaluation on different datasets shows that signatures derived from the consensus signature reveal a much higher stability than signatures learned from all probesets on a microarray, while at the same time being at least as predictive. Furthermore, they are clearly interpretable in terms of enriched pathways, disease associated genes and known drug targets. In summary we thus believe that network based consensus signatures are not only a way to relate seemingly different gene signatures to each other in a functional manner, but also to establish prior knowledge for highly stable and interpretable predictive biomarkers.
基于基因表达谱的癌症诊断和预后生物标志物被视为迈向更好的个性化医疗的重要一步。在过去的十年中,已经发表了许多使用各种计算方法的研究。然而,当比较针对相关临床问题的不同基因特征时,通常只观察到很小的重叠。这可能有多种原因,例如平台的技术差异、生物样本或其在实验室中的处理差异,或由于数据的高维度与小样本量相结合而导致的统计原因,从而导致基因选择不稳定。总之,从生物学角度来看,检索到的基因特征往往难以解释。我们在这里证明,通过将它们映射到蛋白质相互作用网络上,从一组看似不同的基因特征构建共识特征是可能的。通过我们开发的算法确定的密切基因产物的上游常见蛋白,在过表达的 KEGG 途径、疾病相关基因和已知药物靶标方面显示出非常清晰和显著的功能解释。此外,我们表明,这样的共识特征可以作为乳腺癌中预测性生物标志物发现的先验知识。在不同数据集上的评估表明,与从微阵列上的所有探针集学习得到的特征相比,从共识特征派生的特征具有更高的稳定性,而同时至少具有相同的预测能力。此外,它们在富集途径、疾病相关基因和已知药物靶标方面具有明显的可解释性。总之,我们因此相信,基于网络的共识特征不仅是一种以功能方式将看似不同的基因特征相互关联的方法,而且是建立高度稳定和可解释的预测性生物标志物的先验知识的方法。