Department of Chemistry and Biochemistry and Center for Theoretical Biological Physics, University of California at San Diego, La Jolla, California 92093, United States.
Biochemistry. 2011 Dec 6;50(48):10530-9. doi: 10.1021/bi201481a. Epub 2011 Nov 10.
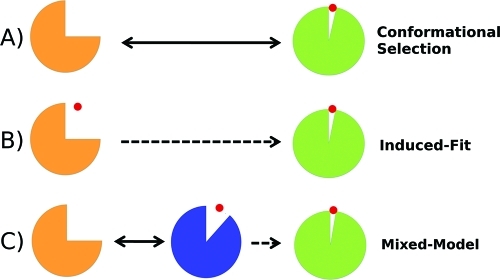
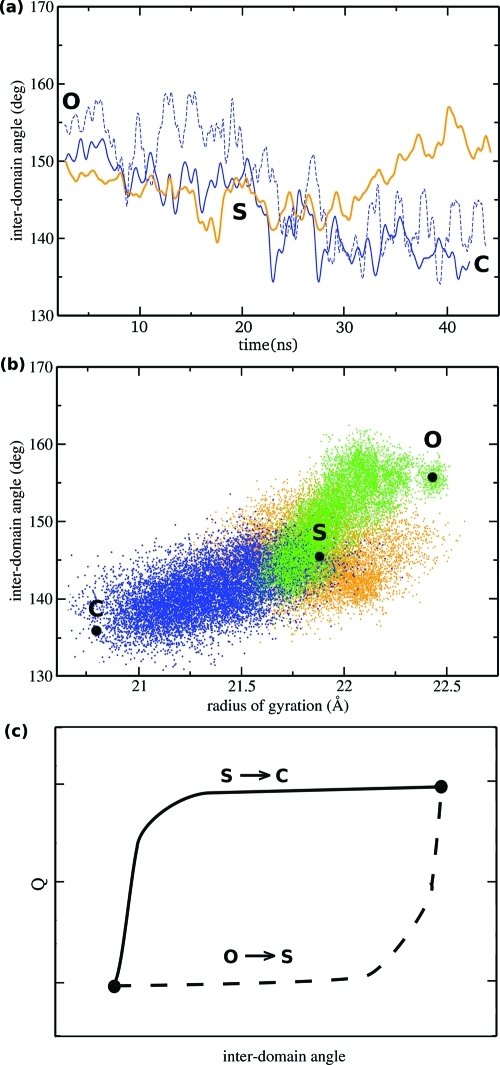
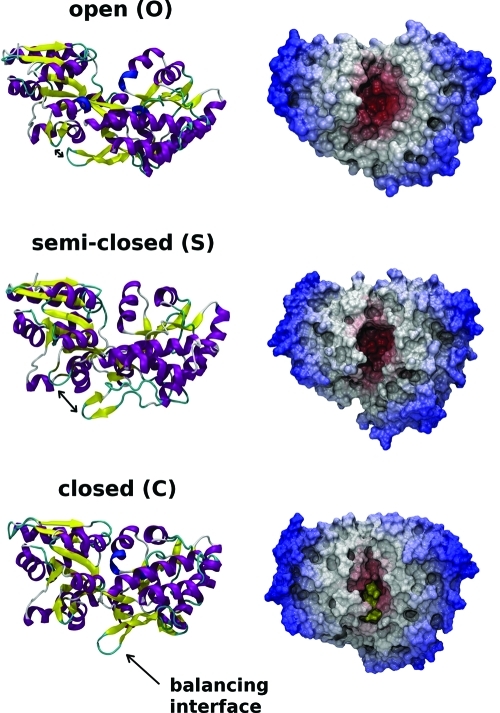
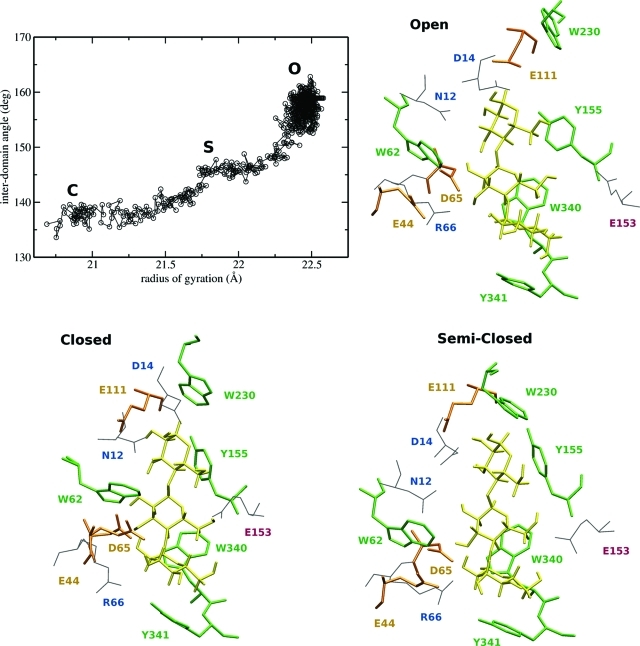
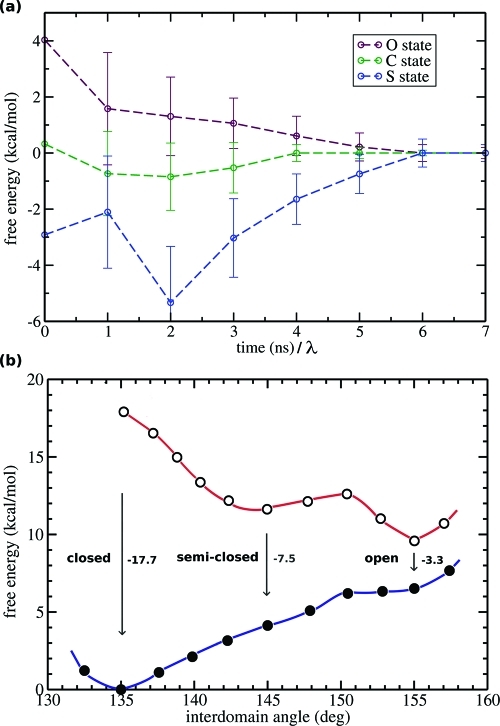
A full characterization of the thermodynamic forces underlying ligand-associated conformational changes in proteins is essential for understanding and manipulating diverse biological processes, including transport, signaling, and enzymatic activity. Recent experiments on the maltose binding protein (MBP) have provided valuable data about the different conformational states implicated in the ligand recognition process; however, a complete picture of the accessible pathways and the associated changes in free energy remains elusive. Here we describe results from advanced accelerated molecular dynamics (aMD) simulations, coupled with adaptively biased force (ABF) and thermodynamic integration (TI) free energy methods. The combination of approaches allows us to track the ligand recognition process on the microsecond time scale and provides a detailed characterization of the protein's dynamic and the relative energy of stable states. We find that an induced-fit (IF) mechanism is most likely and that a mechanism involving both a conformational selection (CS) step and an IF step is also possible. The complete recognition process is best viewed as a "Pac Man" type action where the ligand is initially localized to one domain and naturally occurring hinge-bending vibrations in the protein are able to assist the recognition process by increasing the chances of a favorable encounter with side chains on the other domain, leading to a population shift. This interpretation is consistent with experiments and provides new insight into the complex recognition mechanism. The methods employed here are able to describe IF and CS effects and provide formally rigorous means of computing free energy changes. As such, they are superior to conventional MD and flexible docking alone and hold great promise for future development and applications to drug discovery.
对蛋白质中配体相关构象变化背后的热力学力进行全面描述,对于理解和操纵包括运输、信号传递和酶活性在内的各种生物过程至关重要。最近关于麦芽糖结合蛋白(MBP)的实验提供了有关配体识别过程中涉及的不同构象状态的有价值的数据;然而,关于可及途径和相关自由能变化的完整图景仍然难以捉摸。在这里,我们描述了先进加速分子动力学(aMD)模拟的结果,这些模拟与自适应偏置力(ABF)和热力学积分(TI)自由能方法相结合。这些方法的结合使我们能够在微秒时间尺度上跟踪配体识别过程,并对蛋白质的动态和稳定状态的相对能量进行详细描述。我们发现,诱导契合(IF)机制最有可能,并且涉及构象选择(CS)步骤和 IF 步骤的机制也是可能的。完整的识别过程最好被视为“吃豆人”类型的动作,其中配体最初定位于一个结构域,并且蛋白质中自然发生的铰链弯曲振动能够通过增加与另一个结构域侧链有利相遇的机会来辅助识别过程,从而导致种群转移。这种解释与实验一致,并为复杂的识别机制提供了新的见解。这里使用的方法能够描述 IF 和 CS 效应,并提供正式严格的计算自由能变化的方法。因此,它们优于传统的 MD 和灵活对接单独使用,并且在未来的药物发现发展和应用中具有很大的前景。