School of Pharmacy, Kaohsiung Medical University, Kaohsiung 807, Taiwan.
BMC Bioinformatics. 2011 Nov 15;12:446. doi: 10.1186/1471-2105-12-446.
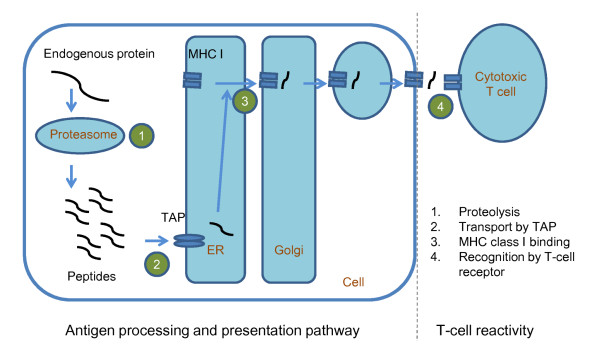
Accurate prediction of peptide immunogenicity and characterization of relation between peptide sequences and peptide immunogenicity will be greatly helpful for vaccine designs and understanding of the immune system. In contrast to the prediction of antigen processing and presentation pathway, the prediction of subsequent T-cell reactivity is a much harder topic. Previous studies of identifying T-cell receptor (TCR) recognition positions were based on small-scale analyses using only a few peptides and concluded different recognition positions such as positions 4, 6 and 8 of peptides with length 9. Large-scale analyses are necessary to better characterize the effect of peptide sequence variations on T-cell reactivity and design predictors of a peptide's T-cell reactivity (and thus immunogenicity). The identification and characterization of important positions influencing T-cell reactivity will provide insights into the underlying mechanism of immunogenicity.
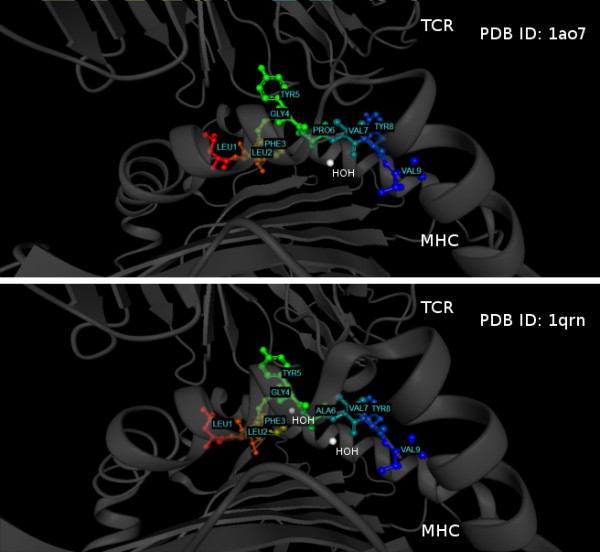
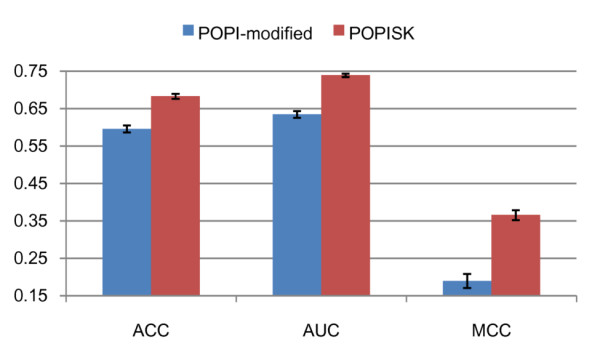
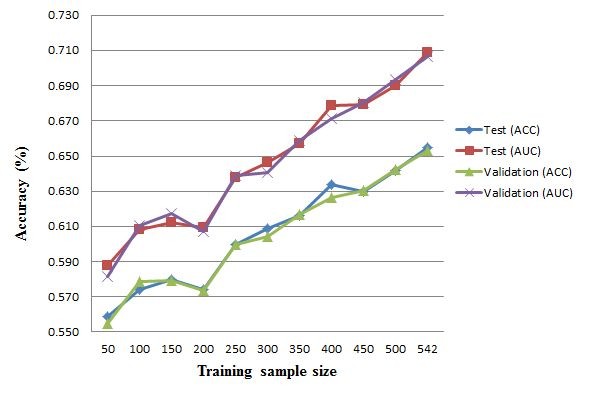
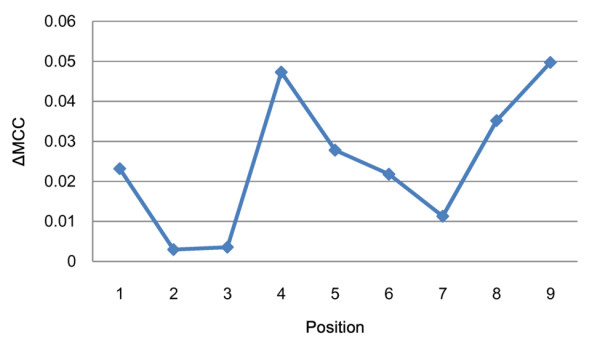
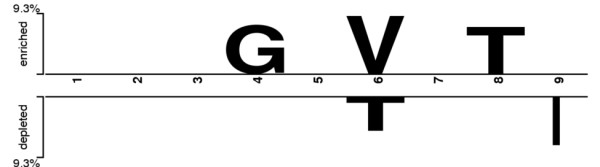
This work establishes a large dataset by collecting immunogenicity data from three major immunology databases. In order to consider the effect of MHC restriction, peptides are classified by their associated MHC alleles. Subsequently, a computational method (named POPISK) using support vector machine with a weighted degree string kernel is proposed to predict T-cell reactivity and identify important recognition positions. POPISK yields a mean 10-fold cross-validation accuracy of 68% in predicting T-cell reactivity of HLA-A2-binding peptides. POPISK is capable of predicting immunogenicity with scores that can also correctly predict the change in T-cell reactivity related to point mutations in epitopes reported in previous studies using crystal structures. Thorough analyses of the prediction results identify the important positions 4, 6, 8 and 9, and yield insights into the molecular basis for TCR recognition. Finally, we relate this finding to physicochemical properties and structural features of the MHC-peptide-TCR interaction.
A computational method POPISK is proposed to predict immunogenicity with scores which are useful for predicting immunogenicity changes made by single-residue modifications. The web server of POPISK is freely available at http://iclab.life.nctu.edu.tw/POPISK.
准确预测肽的免疫原性,并阐明肽序列与肽免疫原性之间的关系,将极大地有助于疫苗设计和免疫系统的理解。与抗原加工和呈递途径的预测相比,后续 T 细胞反应性的预测是一个更加困难的课题。以前的研究确定 T 细胞受体(TCR)识别位置是基于使用少数肽进行小规模分析的,得出的结论是不同的识别位置,例如 9 肽的第 4、6 和 8 位。进行大规模分析对于更好地描述肽序列变化对 T 细胞反应性的影响以及设计预测肽 T 细胞反应性(因此免疫原性)的指标是必要的。确定和描述影响 T 细胞反应性的重要位置将为免疫原性的潜在机制提供深入的了解。
本工作通过从三个主要免疫学数据库收集免疫原性数据,建立了一个大型数据集。为了考虑 MHC 限制的影响,根据相关 MHC 等位基因对肽进行分类。随后,提出了一种使用支持向量机和加权度字符串核的计算方法(命名为 POPISK),用于预测 T 细胞反应性和识别重要识别位置。POPISK 在预测 HLA-A2 结合肽的 T 细胞反应性方面,平均 10 倍交叉验证准确率为 68%。POPISK 能够预测免疫原性,其评分也可以正确预测以前使用晶体结构报告的表位中单个残基突变引起的 T 细胞反应性变化。对预测结果的深入分析确定了重要的第 4、6、8 和 9 位,并深入了解 TCR 识别的分子基础。最后,我们将这一发现与 MHC-肽-TCR 相互作用的物理化学性质和结构特征联系起来。
提出了一种计算方法 POPISK 来预测免疫原性,其评分可用于预测单个残基修饰引起的免疫原性变化。POPISK 的网络服务器可免费在 http://iclab.life.nctu.edu.tw/POPISK 获得。