Malaria and Vector Research Group, Biotechnology Research Center, Pasteur Institute of Iran, Tehran, Iran.
PLoS One. 2011;6(12):e28484. doi: 10.1371/journal.pone.0028484. Epub 2011 Dec 6.
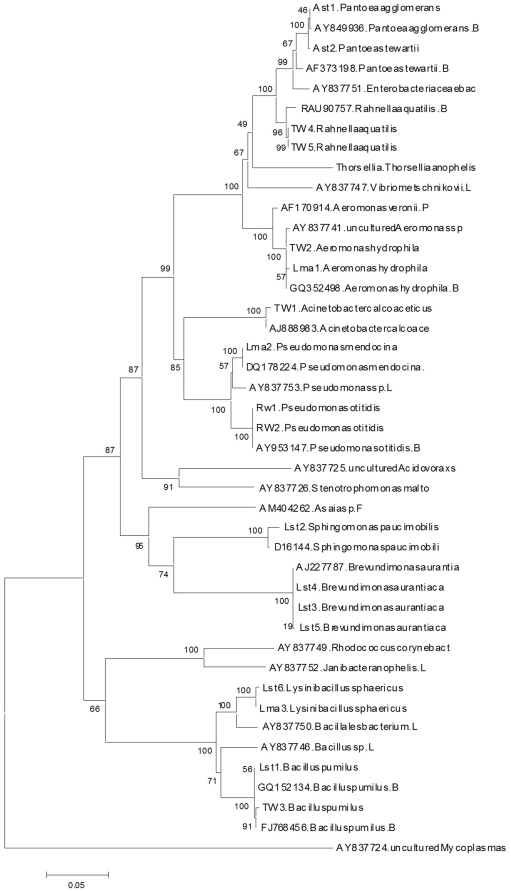
The midgut microbiota associated with Anopheles stephensi and Anopheles maculipennis (Diptera: Culicidae) was investigated for development of a paratransgenesis-based approach to control malaria transmission in Eastern Mediterranean Region (EMR). Here, we present the results of a polymerase chain reaction (PCR) and biochemical-based approaches to identify the female adult and larvae mosquitoe microbiota of these two major malaria vectors, originated from South Eastern and North of Iran. Plating the mosquito midgut contents from lab-reared and field-collected Anopheles spp. was used for microbiota isolation. The gram-negative and gram-positive bacterial colonies were identified by Gram staining and specific mediums. Selected colonies were identified by differential biochemical tests and 16S rRNA gene sequence analysis. A number of 10 An. stephensi and 32 An. maculipennis adult mosquitoes and 15 An. stephensi and 7 An. maculipennis larvae were analyzed and 13 sequences of 16S rRNA gene bacterial species were retrieved, that were categorized in 3 classes and 8 families. The majority of the identified bacteria were belonged to the γ-proteobacteria class, including Pseudomonas sp. and Aeromonas sp. and the others were some closely related to those found in other vector mosquitoes, including Pantoea, Acinetobacter, Brevundimonas, Bacillus, Sphingomonas, Lysinibacillus and Rahnella. The 16S rRNA sequences in the current study aligned with the reference strains available in GenBank were used for construction of the phylogenetic tree that revealed the relatedness among the bacteria identified. The presented data strongly encourage further investigations, to verify the potential role of the detected bacteria for the malaria control in Iran and neighboring countries.
本研究旨在调查与淡色库蚊(Anopheles stephensi)和致倦库蚊(Anopheles maculipennis)(双翅目:蚊科)相关的中肠微生物群,以期开发一种基于共生菌的方法来控制东地中海区域(EMR)的疟疾传播。本研究采用聚合酶链反应(PCR)和基于生化的方法,鉴定了这两种主要疟疾媒介蚊雌成虫和幼虫的微生物群,这些蚊子源自伊朗东南部和北部。通过平板培养法从实验室饲养和野外采集的按蚊肠道内容物中分离微生物群。通过革兰氏染色和特定培养基鉴定革兰氏阴性和革兰氏阳性细菌菌落。通过差异生化试验和 16S rRNA 基因序列分析鉴定选定的菌落。分析了 10 只淡色库蚊和 32 只致倦库蚊成虫以及 15 只淡色库蚊和 7 只致倦库蚊幼虫,共获得 13 个 16S rRNA 基因细菌种序列,可分为 3 个纲和 8 个科。鉴定出的大多数细菌属于γ-变形菌纲,包括假单胞菌属和气单胞菌属,其余与其他媒介蚊中发现的细菌密切相关,包括泛菌属、不动杆菌属、短小杆菌属、芽孢杆菌属、鞘氨醇单胞菌属、粘质沙雷氏菌属和拉恩氏菌属。本研究中 16S rRNA 序列与 GenBank 中可用的参考菌株进行比对,用于构建系统发育树,揭示了所鉴定细菌之间的亲缘关系。目前的研究数据强烈鼓励进一步的研究,以验证所检测到的细菌在伊朗及邻国控制疟疾中的潜在作用。