Translational Genomics Research Institute (TGen), Pathogen Genomics Division, Flagstaff, Arizona, USA.
mBio. 2012 Feb 21;3(1). doi: 10.1128/mBio.00305-11. Print 2012.
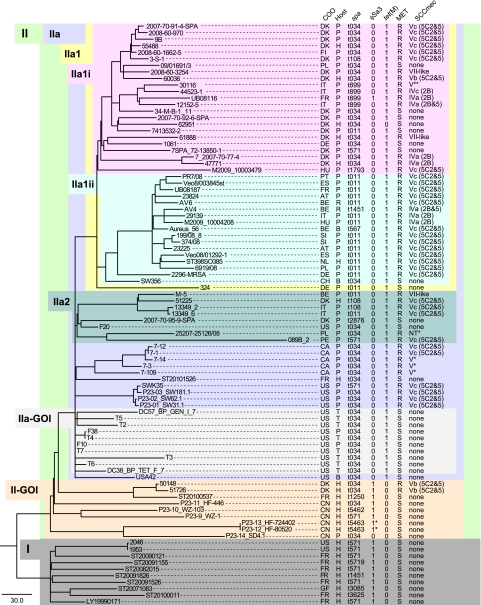
Since its discovery in the early 2000s, methicillin-resistant Staphylococcus aureus (MRSA) clonal complex 398 (CC398) has become a rapidly emerging cause of human infections, most often associated with livestock exposure. We applied whole-genome sequence typing to characterize a diverse collection of CC398 isolates (n = 89), including MRSA and methicillin-susceptible S. aureus (MSSA) from animals and humans spanning 19 countries and four continents. We identified 4,238 single nucleotide polymorphisms (SNPs) among the 89 core genomes. Minimal homoplasy (consistency index = 0.9591) was detected among parsimony-informative SNPs, allowing for the generation of a highly accurate phylogenetic reconstruction of the CC398 clonal lineage. Phylogenetic analyses revealed that MSSA from humans formed the most ancestral clades. The most derived lineages were composed predominantly of livestock-associated MRSA possessing three different staphylococcal cassette chromosome mec element (SCCmec) types (IV, V, and VII-like) including nine subtypes. The human-associated isolates from the basal clades carried phages encoding human innate immune modulators that were largely missing among the livestock-associated isolates. Our results strongly suggest that livestock-associated MRSA CC398 originated in humans as MSSA. The lineage appears to have undergone a rapid radiation in conjunction with the jump from humans to livestock, where it subsequently acquired tetracycline and methicillin resistance. Further analyses are required to estimate the number of independent genetic events leading to the methicillin-resistant sublineages, but the diversity of SCCmec subtypes is suggestive of strong and diverse antimicrobial selection associated with food animal production.
Modern food animal production is characterized by densely concentrated animals and routine antibiotic use, which may facilitate the emergence of novel antibiotic-resistant zoonotic pathogens. Our findings strongly support the idea that livestock-associated MRSA CC398 originated as MSSA in humans. The jump of CC398 from humans to livestock was accompanied by the loss of phage-carried human virulence genes, which likely attenuated its zoonotic potential, but it was also accompanied by the acquisition of tetracycline and methicillin resistance. Our findings exemplify a bidirectional zoonotic exchange and underscore the potential public health risks of widespread antibiotic use in food animal production.
自 21 世纪初发现以来,耐甲氧西林金黄色葡萄球菌(MRSA)克隆复合体 398(CC398)已迅速成为人类感染的一个主要原因,大多数情况下与接触家畜有关。我们应用全基因组序列分型来对包括来自动物和人类的 19 个国家和四个大陆的 89 株 CC398 分离株(MRSA 和甲氧西林敏感金黄色葡萄球菌(MSSA))进行了多样化的特征描述。我们在 89 个核心基因组中鉴定出 4238 个单核苷酸多态性(SNP)。简约信息 SNP 之间的同源性最小(一致性指数=0.9591),允许对 CC398 克隆谱系进行高度准确的系统发育重建。系统发育分析表明,来自人类的 MSSA 形成了最古老的分支。最衍生的谱系主要由具有三种不同葡萄球菌盒染色体 mec 元件(SCCmec)类型(IV、V 和 VII 样)的畜群相关 MRSA 组成,包括九个亚型。来自基础分支的人类相关分离株携带编码人类先天免疫调节剂的噬菌体,而这些噬菌体在畜群相关分离株中大多缺失。我们的研究结果强烈表明,与家畜相关的 MRSA CC398 起源于人类 MSSA。该谱系似乎与从人类到家畜的跳跃同时发生了快速辐射,随后在那里获得了四环素和甲氧西林耐药性。需要进一步分析以估计导致耐甲氧西林亚谱系的独立遗传事件的数量,但 SCCmec 亚型的多样性表明与食品动物生产相关的强烈和多样化的抗菌选择。
现代食品动物生产的特点是动物高度集中和常规使用抗生素,这可能促进新型抗生素耐药性人畜共患病病原体的出现。我们的研究结果强烈支持这样一种观点,即与家畜相关的 MRSA CC398 起源于人类 MSSA。CC398 从人类跳跃到家畜,同时丢失了噬菌体携带的人类毒力基因,这可能削弱了其人畜共患潜力,但也同时获得了四环素和甲氧西林耐药性。我们的研究结果例证了双向人畜共患病的交换,并强调了在食品动物生产中广泛使用抗生素的潜在公共卫生风险。