McCormick Genomic and Proteomics Center, The George Washington University, Washington, DC 20037, USA.
Sci Rep. 2012;2:264. doi: 10.1038/srep00264. Epub 2012 Feb 14.
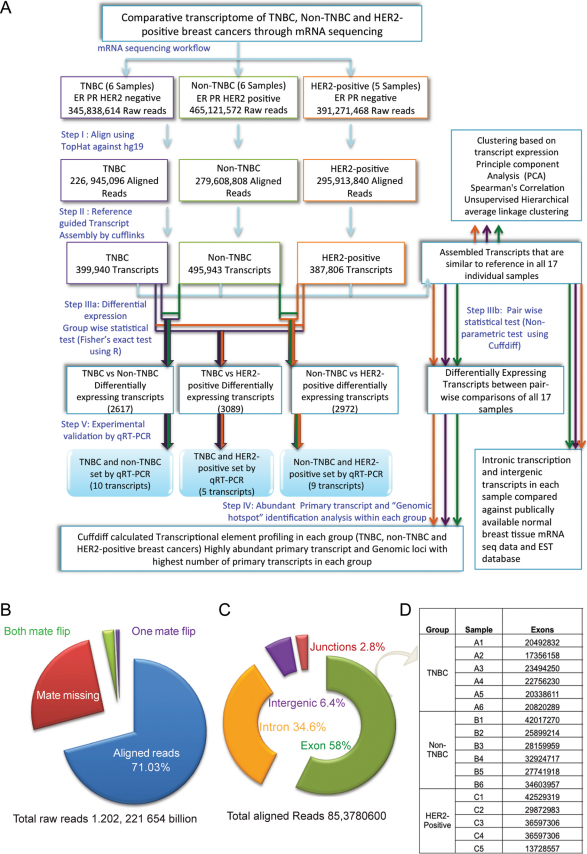
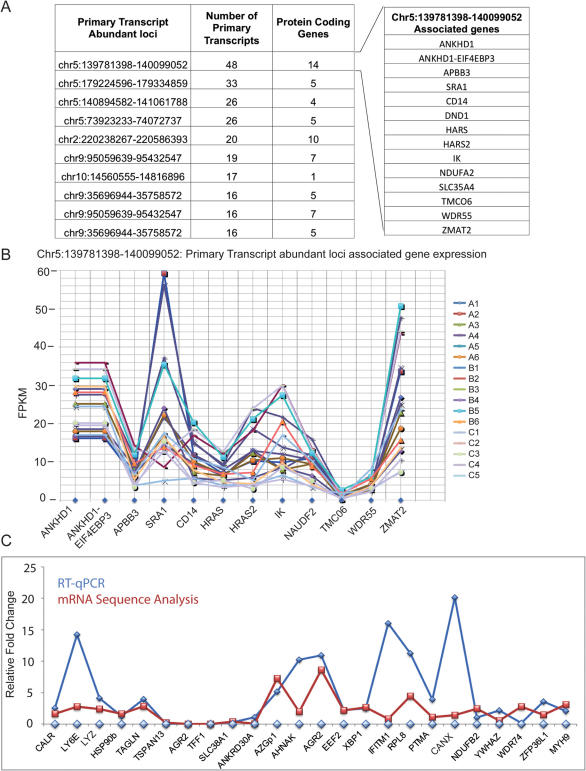
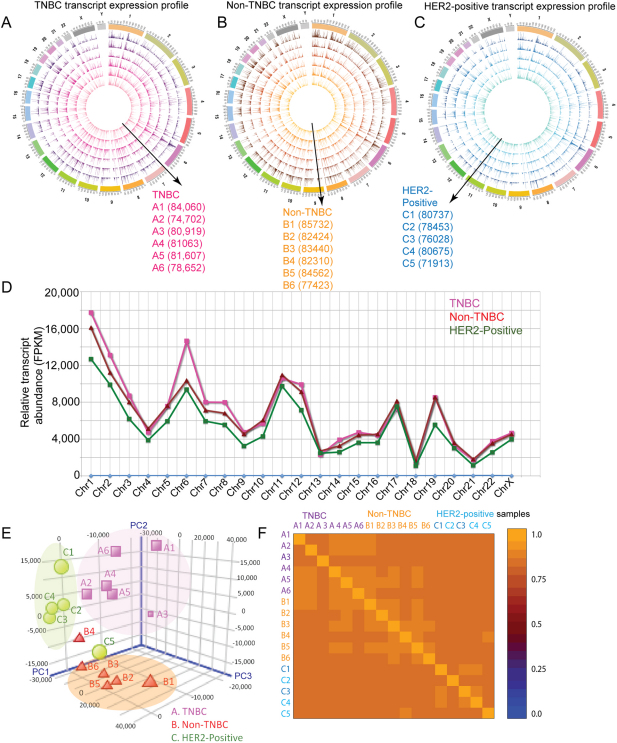
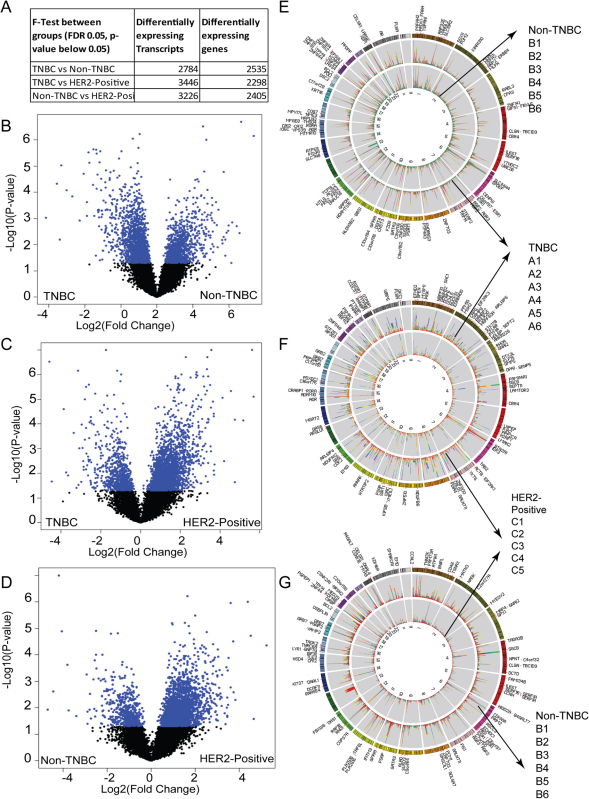
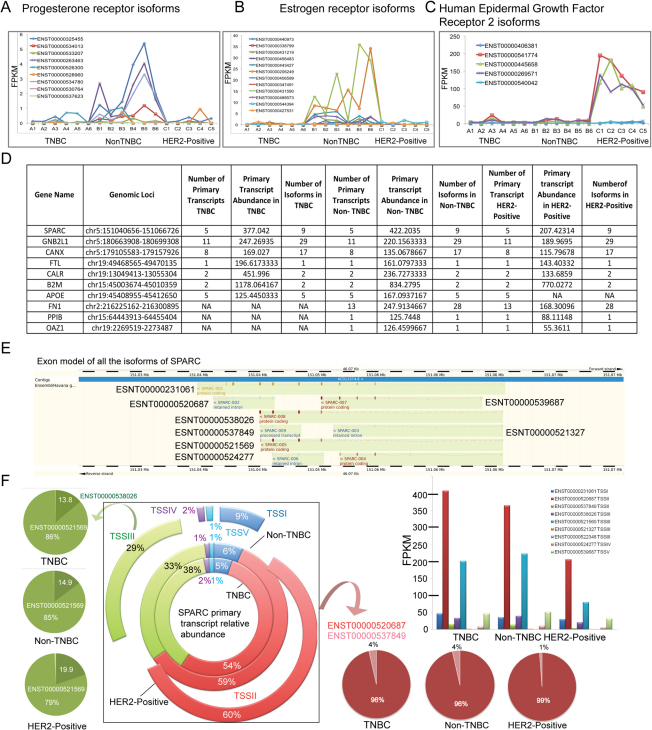
Breast cancer is a heterogeneous disease with a poorly defined genetic landscape, which poses a major challenge in diagnosis and treatment. By massively parallel mRNA sequencing, we obtained 1.2 billion reads from 17 individual human tissues belonging to TNBC, Non-TNBC, and HER2-positive breast cancers and defined their comprehensive digital transcriptome for the first time. Surprisingly, we identified a high number of novel and unannotated transcripts, revealing the global breast cancer transcriptomic adaptations. Comparative transcriptomic analyses elucidated differentially expressed transcripts between the three breast cancer groups, identifying several new modulators of breast cancer. Our study also identified common transcriptional regulatory elements, such as highly abundant primary transcripts, including osteonectin, RACK1, calnexin, calreticulin, FTL, and B2M, and "genomic hotspots" enriched in primary transcripts between the three groups. Thus, our study opens previously unexplored niches that could enable a better understanding of the disease and the development of potential intervention strategies.
乳腺癌是一种异质性疾病,其遗传图谱定义不明确,这对诊断和治疗构成了重大挑战。通过大规模平行 mRNA 测序,我们从属于三阴性乳腺癌、非三阴性乳腺癌和 HER2 阳性乳腺癌的 17 个人类组织中获得了 12 亿个读数,并首次对其进行了全面的数字转录组定义。令人惊讶的是,我们鉴定了大量新的未注释的转录本,揭示了全球乳腺癌转录组的适应性。比较转录组分析阐明了三组乳腺癌之间差异表达的转录本,鉴定了几种新的乳腺癌调节剂。我们的研究还鉴定了常见的转录调控元件,例如高度丰富的初级转录本,包括骨粘连蛋白、RACK1、钙网蛋白、钙结合蛋白、FTL 和 B2M,以及三组之间富含初级转录本的“基因组热点”。因此,我们的研究开辟了以前未探索的领域,这可能有助于更好地理解疾病并开发潜在的干预策略。