Key Laboratory of Systems Biology, SIBS-Novo Nordisk Translational Research Centre for PreDiabetes, Shanghai Institutes for Biological Sciences, Chinese Academy of Sciences, Shanghai 200031, China.
BMC Bioinformatics. 2012 May 8;13 Suppl 7(Suppl 7):S6. doi: 10.1186/1471-2105-13-S7-S6.
Mycobacterium tuberculosis is an infectious bacterium posing serious threats to human health. Due to the difficulty in performing molecular biology experiments to detect protein interactions, reconstruction of a protein interaction map of M. tuberculosis by computational methods will provide crucial information to understand the biological processes in the pathogenic microorganism, as well as provide the framework upon which new therapeutic approaches can be developed.
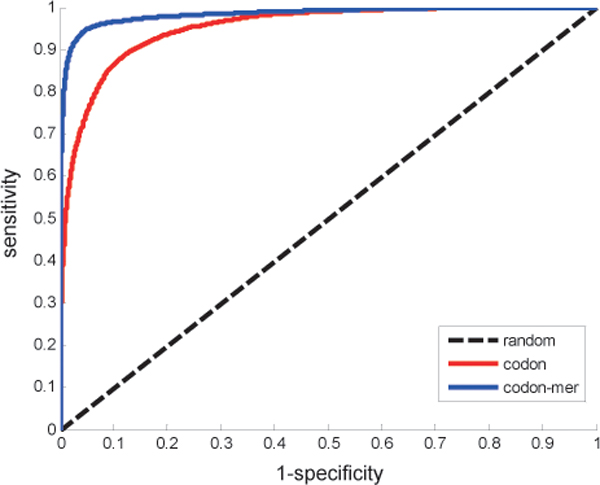
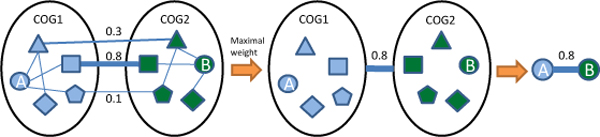
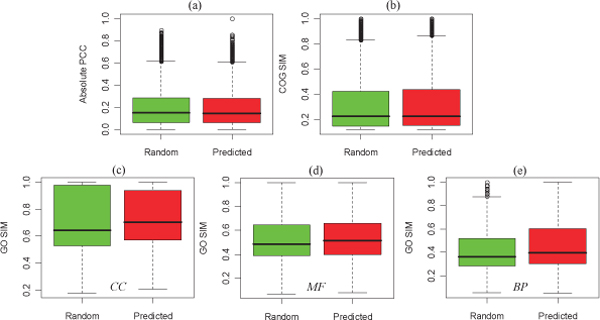
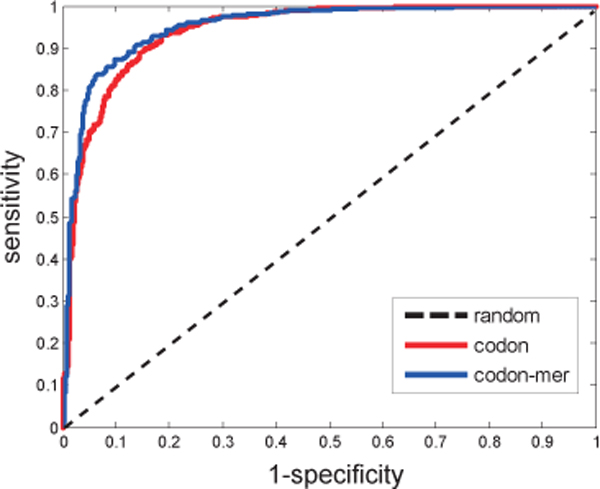
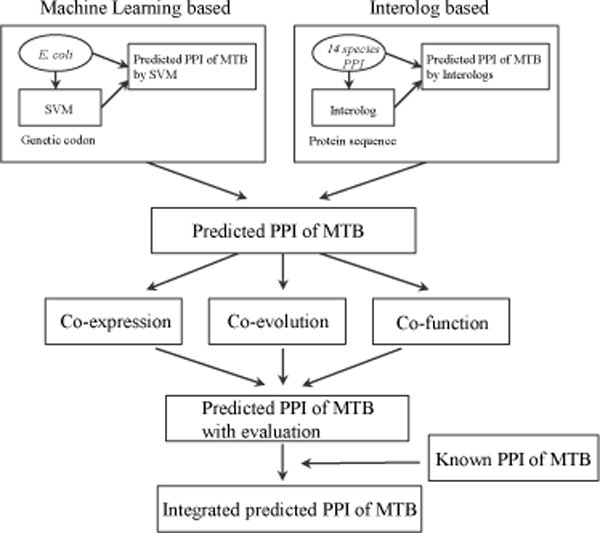
In this paper, we constructed an integrated M. tuberculosis protein interaction network by machine learning and ortholog-based methods. Firstly, we built a support vector machine (SVM) method to infer the protein interactions of M. tuberculosis H37Rv by gene sequence information. We tested our predictors in Escherichia coli and mapped the genetic codon features underlying its protein interactions to M. tuberculosis. Moreover, the documented interactions of 14 other species were mapped to the interactome of M. tuberculosis by the interolog method. The ensemble protein interactions were validated by various functional relationships, i.e., gene coexpression, evolutionary relationship and functional similarity, extracted from heterogeneous data sources. The accuracy and validation demonstrate the effectiveness and efficiency of our framework.
A protein interaction map of M. tuberculosis is inferred from genetic codons and interologs. The prediction accuracy and numerically experimental validation demonstrate the effectiveness and efficiency of our method. Furthermore, our methods can be straightforwardly extended to infer the protein interactions of other bacterial species.
结核分枝杆菌是一种传染性细菌,对人类健康构成严重威胁。由于难以进行分子生物学实验来检测蛋白质相互作用,因此通过计算方法重建结核分枝杆菌的蛋白质相互作用图谱将为理解致病微生物中的生物学过程提供关键信息,并为开发新的治疗方法提供框架。
本文通过机器学习和基于同源物的方法构建了一个整合的结核分枝杆菌蛋白质相互作用网络。首先,我们构建了一个支持向量机(SVM)方法,通过基因序列信息来推断结核分枝杆菌 H37Rv 的蛋白质相互作用。我们在大肠杆菌中测试了我们的预测器,并将其蛋白质相互作用的遗传密码特征映射到结核分枝杆菌上。此外,通过同源物方法将 14 种其他物种的已有相互作用映射到结核分枝杆菌的相互作用组中。通过从异构数据源中提取的各种功能关系,即基因共表达、进化关系和功能相似性,对组合蛋白质相互作用进行了验证。准确性和验证表明了我们框架的有效性和效率。
从遗传密码和同源物推断出结核分枝杆菌的蛋白质相互作用图谱。预测准确性和数值实验验证证明了我们方法的有效性和效率。此外,我们的方法可以直接扩展到推断其他细菌物种的蛋白质相互作用。