Department of Medical Oncology and Daniel Den Hoed Cancer Center, Erasmus University Medical Center, Dr. Molewaterplein 50, Rotterdam, the Netherlands.
J Mammary Gland Biol Neoplasia. 2012 Jun;17(2):155-64. doi: 10.1007/s10911-012-9252-6. Epub 2012 May 30.
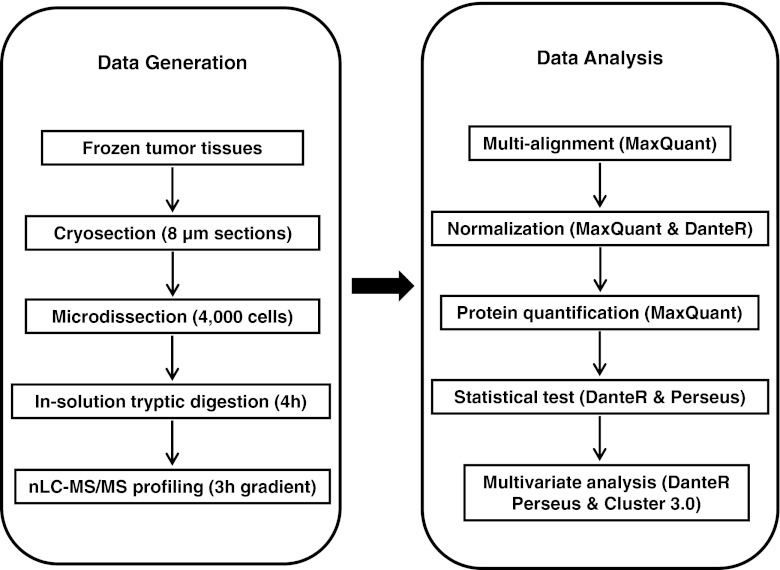
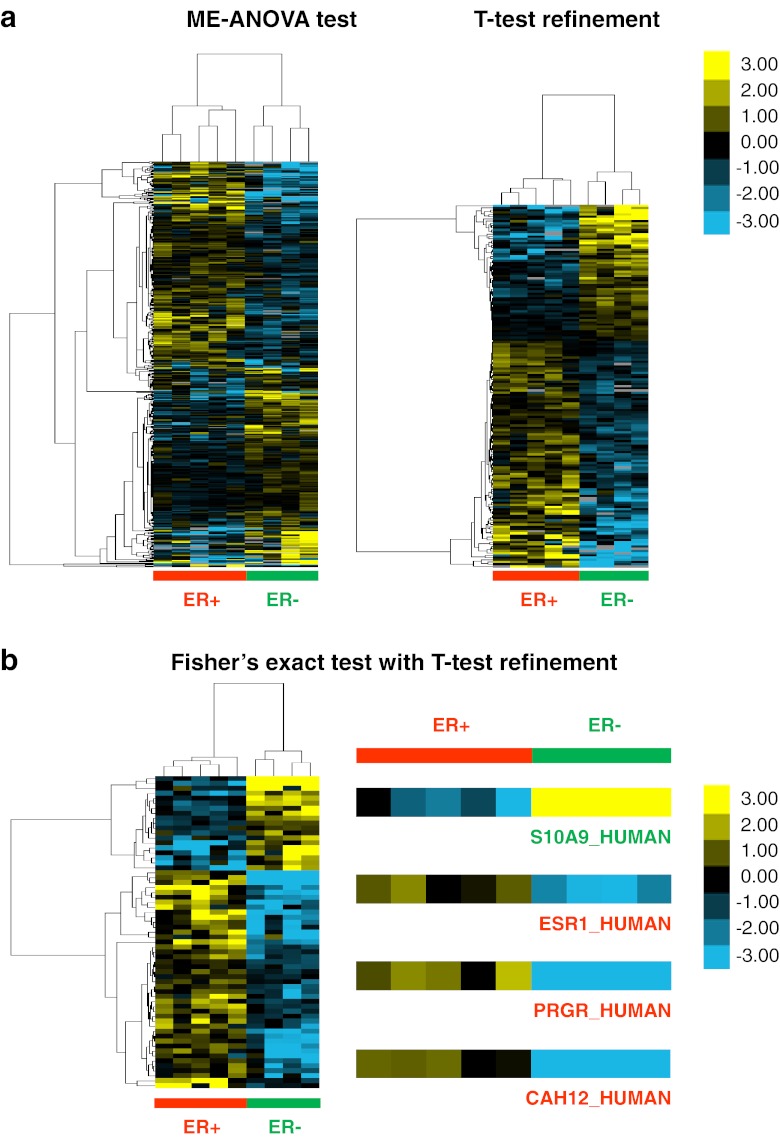
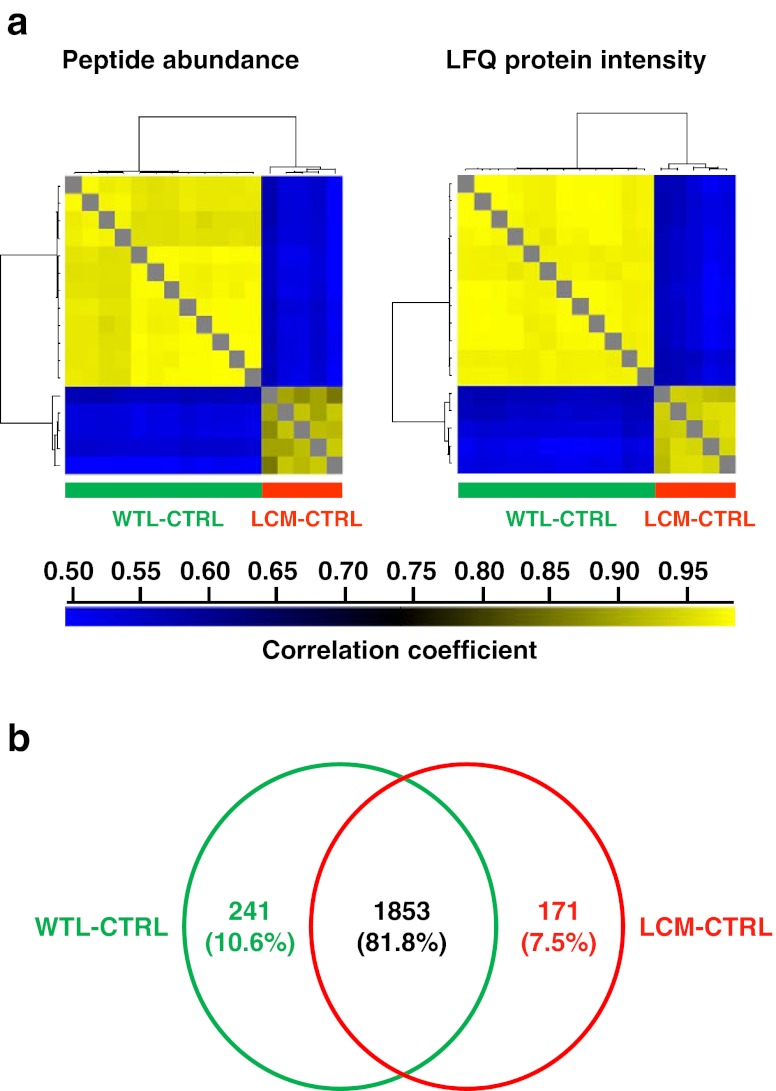
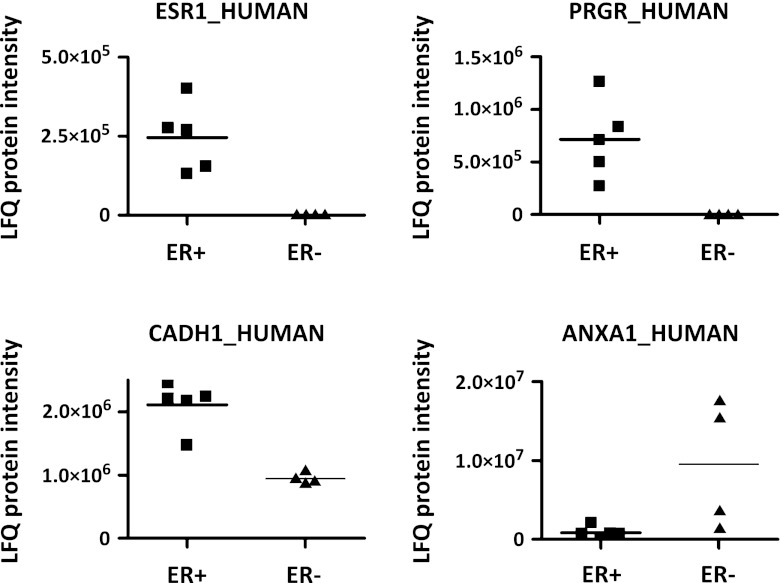
Mass spectrometry (MS)-based label-free proteomics offers an unbiased approach to screen biomarkers related to disease progression and therapy-resistance of breast cancer on the global scale. However, multi-step sample preparation can introduce large variation in generated data, while inappropriate statistical methods will lead to false positive hits. All these issues have hampered the identification of reliable protein markers. A workflow, which integrates reproducible and robust sample preparation and data handling methods, is highly desirable in clinical proteomics investigations. Here we describe a label-free tissue proteomics pipeline, which encompasses laser capture microdissection (LCM) followed by nanoscale liquid chromatography and high resolution MS. This pipeline routinely identifies on average ∼10,000 peptides corresponding to ∼1,800 proteins from sub-microgram amounts of protein extracted from ∼4,000 LCM breast cancer epithelial cells. Highly reproducible abundance data were generated from different technical and biological replicates. As a proof-of-principle, comparative proteome analysis was performed on estrogen receptor α positive or negative (ER+/-) samples, and commonly known differentially expressed proteins related to ER expression in breast cancer were identified. Therefore, we show that our tissue proteomics pipeline is robust and applicable for the identification of breast cancer specific protein markers.
基于质谱(MS)的无标记蛋白质组学提供了一种在全球范围内筛选与乳腺癌疾病进展和治疗耐药相关的生物标志物的无偏方法。然而,多步样品制备会在产生的数据中引入较大的差异,而不适当的统计方法将导致假阳性结果。所有这些问题都阻碍了可靠蛋白质标记物的识别。在临床蛋白质组学研究中,非常需要集成可重复和稳健的样品制备和数据处理方法的工作流程。在这里,我们描述了一种无标记组织蛋白质组学工作流程,它包括激光捕获显微切割(LCM),然后是纳米液相色谱和高分辨率 MS。该工作流程通常可以从亚微克量的蛋白质中鉴定出约 10000 个肽,对应于约 1800 种蛋白质,这些蛋白质是从约 4000 个 LCM 乳腺癌上皮细胞中提取的。从不同的技术和生物学重复中生成了高度可重复的丰度数据。作为原理验证,对雌激素受体α阳性或阴性(ER +/-)样本进行了比较蛋白质组学分析,鉴定出了与乳腺癌中 ER 表达相关的常见差异表达蛋白。因此,我们表明我们的组织蛋白质组学工作流程是稳健的,适用于鉴定乳腺癌特异性蛋白质标记物。