Department of Computer Science and Engineering, Ohio State University, Columbus, OH, USA.
Bioinformatics. 2012 Jun 15;28(12):i49-58. doi: 10.1093/bioinformatics/bts212.
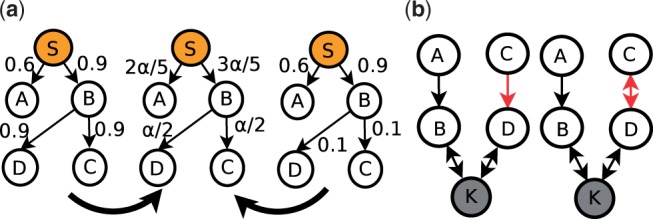
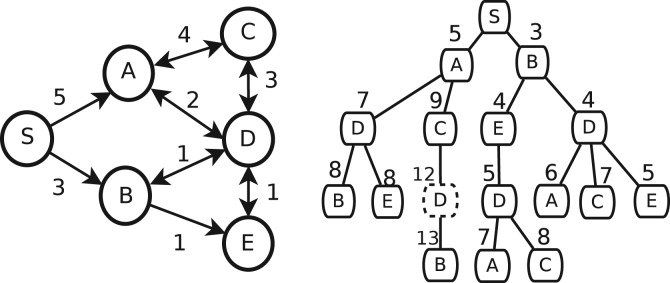
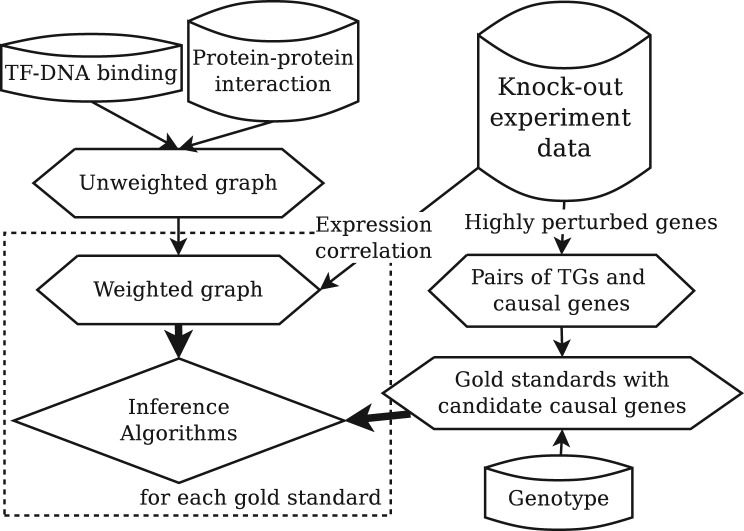
Inferring the underlying regulatory pathways within a gene interaction network is a fundamental problem in Systems Biology to help understand the complex interactions and the regulation and flow of information within a system-of-interest. Given a weighted gene network and a gene in this network, the goal of an inference algorithm is to identify the potential regulatory pathways passing through this gene.
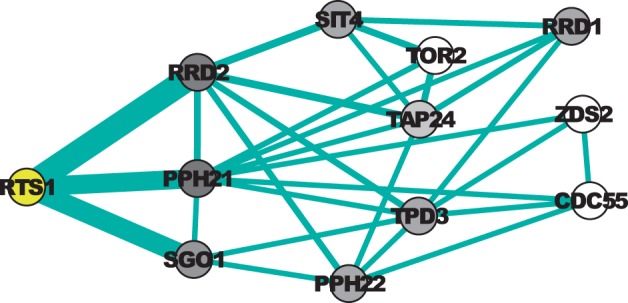
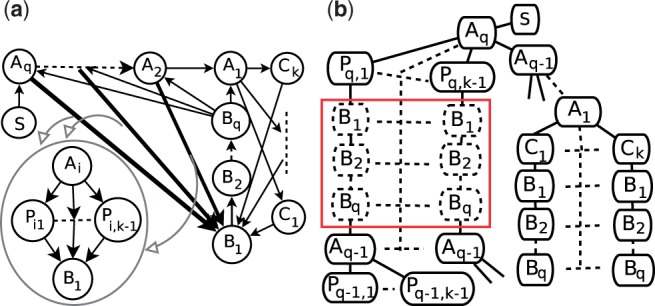
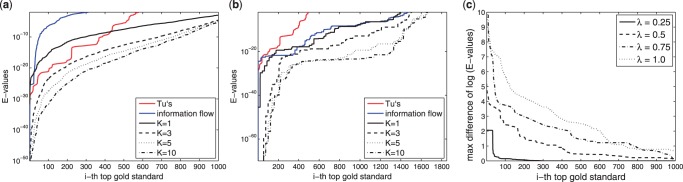
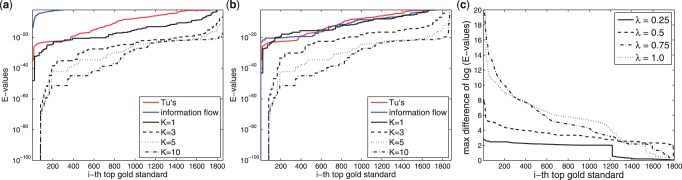
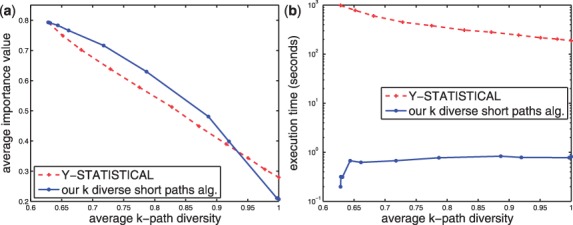
In a departure from previous approaches that largely rely on the random walk model, we propose a novel single-source k-shortest paths based algorithm to address this inference problem. An important element of our approach is to explicitly account for and enhance the diversity of paths discovered by our algorithm. The intuition here is that diversity in paths can help enrich different functions and thereby better position one to understand the underlying system-of-interest. Results on the yeast gene network demonstrate the utility of the proposed approach over extant state-of-the-art inference algorithms. Beyond utility, our algorithm achieves a significant speedup over these baselines.
All data and codes are freely available upon request.
在系统生物学中,推断基因交互网络中的潜在调控途径是一个基本问题,有助于理解系统中复杂的相互作用以及信息的调控和流动。给定一个加权基因网络和这个网络中的一个基因,推断算法的目标是识别通过这个基因的潜在调控途径。
与以前主要依赖随机游走模型的方法不同,我们提出了一种新的基于单源 k 条最短路径的算法来解决这个推断问题。我们方法的一个重要元素是明确考虑和增强我们算法发现的路径的多样性。这里的直觉是,路径的多样性可以帮助丰富不同的功能,从而更好地理解潜在的系统。在酵母基因网络上的结果表明,该方法优于现有的最先进的推断算法。除了实用性之外,我们的算法在这些基准测试上实现了显著的加速。
所有数据和代码均可应要求免费提供。