Laboratory for Computational Biology and Bioinformatics, School of Computer and Communication Sciences, École Polytechnique Fédérale de Lausanne (EPFL), Lausanne, Switzerland.
PLoS One. 2012;7(8):e39573. doi: 10.1371/journal.pone.0039573. Epub 2012 Aug 3.
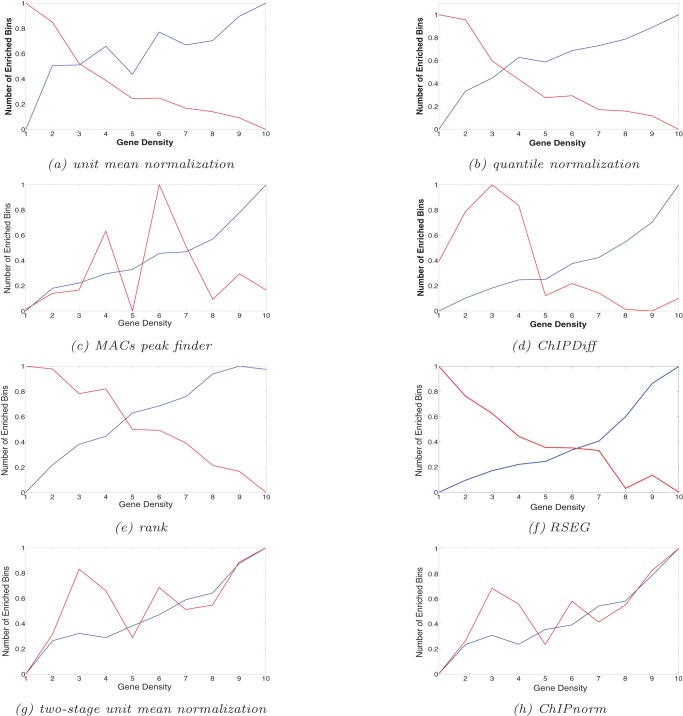
The advent of high-throughput technologies such as ChIP-seq has made possible the study of histone modifications. A problem of particular interest is the identification of regions of the genome where different cell types from the same organism exhibit different patterns of histone enrichment. This problem turns out to be surprisingly difficult, even in simple pairwise comparisons, because of the significant level of noise in ChIP-seq data. In this paper we propose a two-stage statistical method, called ChIPnorm, to normalize ChIP-seq data, and to find differential regions in the genome, given two libraries of histone modifications of different cell types. We show that the ChIPnorm method removes most of the noise and bias in the data and outperforms other normalization methods. We correlate the histone marks with gene expression data and confirm that histone modifications H3K27me3 and H3K4me3 act as respectively a repressor and an activator of genes. Compared to what was previously reported in the literature, we find that a substantially higher fraction of bivalent marks in ES cells for H3K27me3 and H3K4me3 move into a K27-only state. We find that most of the promoter regions in protein-coding genes have differential histone-modification sites. The software for this work can be downloaded from http://lcbb.epfl.ch/software.html.
高通量技术(如 ChIP-seq)的出现使得研究组蛋白修饰成为可能。一个特别有趣的问题是鉴定来自同一生物体的不同细胞类型中组蛋白富集呈现不同模式的基因组区域。由于 ChIP-seq 数据中存在大量噪声,即使在简单的两两比较中,这个问题也变得非常困难。在本文中,我们提出了一种两阶段统计方法,称为 ChIPnorm,用于对 ChIP-seq 数据进行标准化,并在给定来自不同细胞类型的两种组蛋白修饰文库的情况下,找到基因组中的差异区域。我们表明,ChIPnorm 方法可以去除数据中的大部分噪声和偏差,并优于其他标准化方法。我们将组蛋白标记与基因表达数据相关联,并证实组蛋白修饰 H3K27me3 和 H3K4me3 分别作为基因的抑制剂和激活剂。与之前文献中的报道相比,我们发现 ES 细胞中 H3K27me3 和 H3K4me3 的双价标记有很大一部分转变为仅 H3K27 状态。我们发现大多数蛋白质编码基因的启动子区域都有差异的组蛋白修饰位点。这项工作的软件可以从 http://lcbb.epfl.ch/software.html 下载。