Key Laboratory Animal Genetics, Breeding and Reproduction, Ministry of Agriculture, College of Animal Science and Technology, National Engineering Laboratory for Animal Breeding, China Agricultural University, Beijing 100193, China.
BMC Genomics. 2012 Sep 18;13:488. doi: 10.1186/1471-2164-13-488.
Lymphocytes act as a major component of the adaptive immune system, taking very crucial responsibility for immunity. Differences in proportions of T-cell subpopulations in peripheral blood among individuals under same conditions provide evidence of genetic control on these traits, but little is known about the genetic mechanism of them, especially in swine. Identification of the genetic control on these variants may help the genetic improvement of immune capacity through selection.
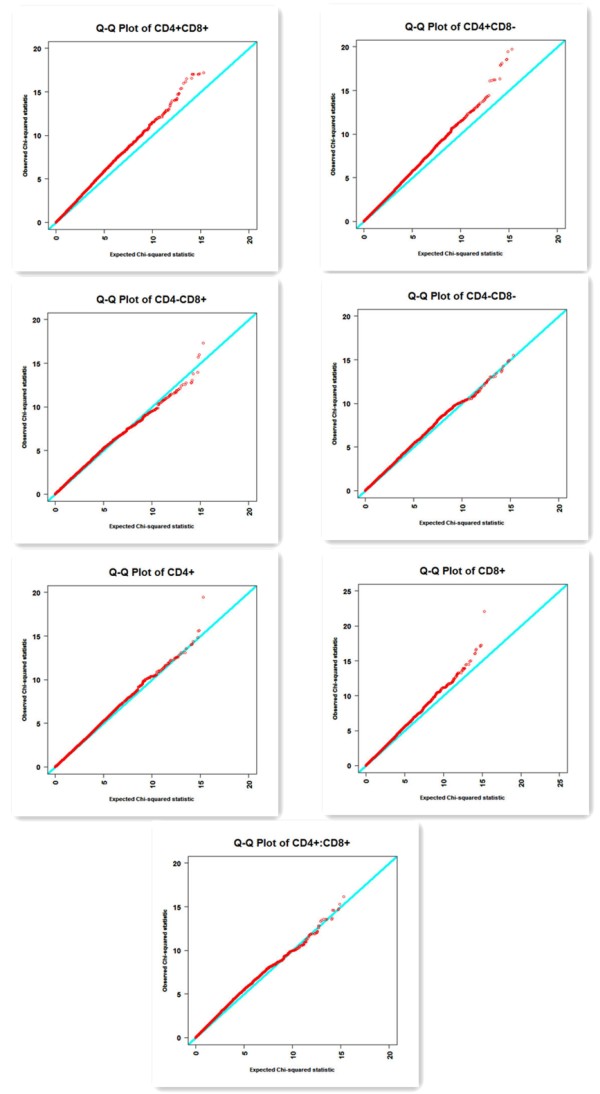
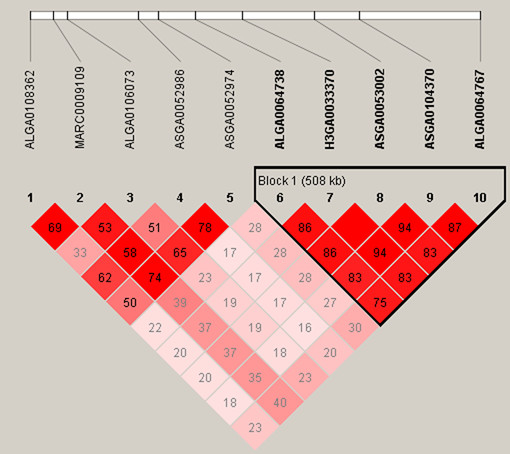
To identify genomic regions responsible for these immune traits in swine, a genome-wide association study was conducted. A total of 675 pigs of three breeds were involved in the study. At 21 days of age, all individuals were vaccinated with modified live classical swine fever vaccine. Blood samples were collected when the piglets were 20 and 35 days of age, respectively. Seven traits, including the proportions of CD4+, CD8+, CD4+CD8+, CD4+CD8-, CD4-CD8+, CD4-CD8- and the ratio of CD4+ to CD8+ T cells were measured at the two ages. All the samples were genotyped for 62,163 single nucleotide polymorphisms (SNP) using the Illumina porcineSNP60k BeadChip. 40833 SNPs were selected after quality control for association tests between SNPs and each immune trait considered based on a single-locus regression model. To tackle the issue of multiple testing in GWAS, 10,000 permutations were performed to determine the chromosome-wise and genome-wise significance levels of association tests. In total, 61 SNPs with chromosome-wise significance level and 3 SNPs with genome-wise significance level were identified. 27 significant SNPs were located within the immune-related QTL regions reported in previous studies. Furthermore, several significant SNPs fell into the regions harboring known immunity-related genes, 14 of them fell into the regions which harbor some known T cell-related genes.
Our study demonstrated that genome-wide association studies would be a feasible way for revealing the potential genetics variants affecting T-cell subpopulations. Results herein lay a preliminary foundation for further identifying the causal mutations underlying swine immune capacity in follow-up studies.
淋巴细胞作为适应性免疫系统的主要组成部分,对免疫起着至关重要的作用。在相同条件下,个体外周血中 T 细胞亚群比例的差异为这些性状提供了遗传控制的证据,但对其遗传机制知之甚少,特别是在猪中。鉴定这些变异的遗传控制可能有助于通过选择来提高免疫能力。
为了鉴定猪中与这些免疫性状相关的基因组区域,进行了全基因组关联研究。该研究共涉及三个品种的 675 头猪。所有个体在 21 日龄时均用改良活经典猪瘟疫苗进行疫苗接种。分别在仔猪 20 日龄和 35 日龄时采集血样。在这两个年龄分别测量了 7 个性状,包括 CD4+、CD8+、CD4+CD8+、CD4+CD8-、CD4-CD8+、CD4-CD8-和 CD4+与 CD8+T 细胞的比值。使用 Illumina porcineSNP60k BeadChip 对所有样本进行了 62,163 个单核苷酸多态性(SNP)的基因分型。在基于单基因座回归模型对 SNP 与每个免疫性状进行关联测试后,经过质量控制,选择了 40833 个 SNP。为了解决 GWAS 中的多重测试问题,进行了 10,000 次随机排列,以确定染色体和基因组关联测试的显著水平。总共鉴定出 61 个具有染色体显著水平的 SNP 和 3 个具有基因组显著水平的 SNP。27 个显著 SNP 位于之前研究报道的免疫相关 QTL 区域内。此外,一些显著 SNP 落入了包含已知免疫相关基因的区域,其中 14 个 SNP 落入了包含一些已知 T 细胞相关基因的区域。
本研究表明,全基因组关联研究将是揭示影响 T 细胞亚群的潜在遗传变异的可行方法。本研究结果为进一步鉴定后续研究中猪免疫能力的因果突变奠定了初步基础。