Center for Cancer Genomics, Wake Forest University, Winston-Salem, North Carolina, United States of America.
PLoS One. 2012;7(10):e48455. doi: 10.1371/journal.pone.0048455. Epub 2012 Oct 31.
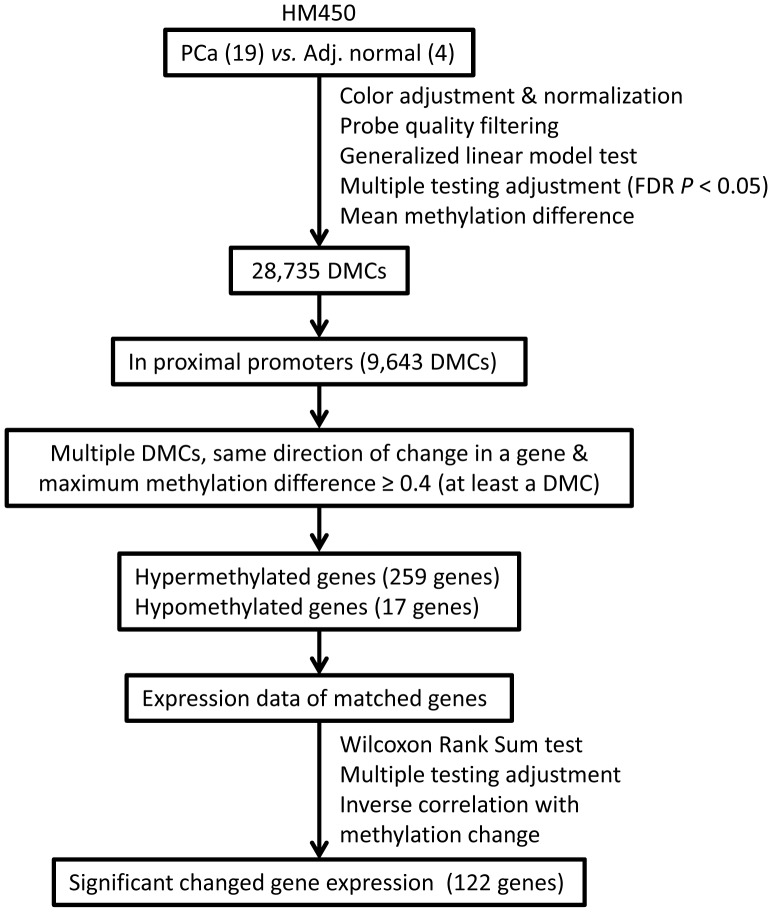
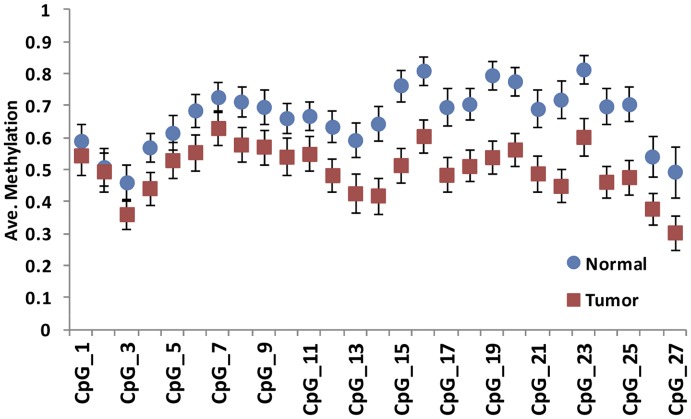
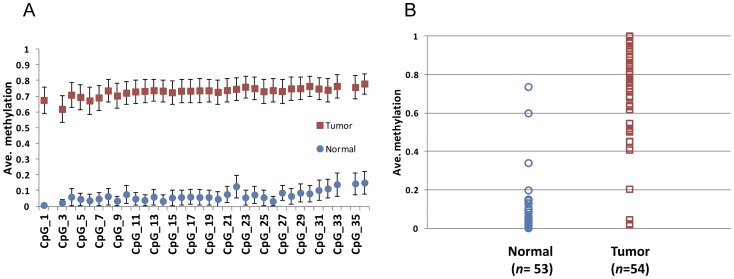
Many differentially methylated genes have been identified in prostate cancer (PCa), primarily using candidate gene-based assays. Recently, several global DNA methylation profiles have been reported in PCa, however, each of these has weaknesses in terms of ability to observe global DNA methylation alterations in PCa. We hypothesize that there remains unidentified aberrant DNA methylation in PCa, which may be identified using higher resolution assay methods. We used the newly developed Illumina HumanMethylation450 BeadChip in PCa (n = 19) and adjacent normal tissues (n = 4) and combined these with gene expression data for identifying new DNA methylation that may have functional consequences in PCa development and progression. We also confirmed our methylation results in an independent data set. Two aberrant DNA methylation genes were validated among an additional 56 PCa samples and 55 adjacent normal tissues. A total 28,735 CpG sites showed significant differences in DNA methylation (FDR adjusted P<0.05), defined as a mean methylation difference of at least 20% between PCa and normal samples. Furthermore, a total of 122 genes had more than one differentially methylated CpG site in their promoter region and a gene expression pattern that was inverse to the direction of change in DNA methylation (e.g. decreased expression with increased methylation, and vice-versa). Aberrant DNA methylation of two genes, AOX1 and SPON2, were confirmed via bisulfate sequencing, with most of the respective CpG sites showing significant differences between tumor samples and normal tissues. The AOX1 promoter region showed hypermethylation in 92.6% of 54 tested PCa samples in contrast to only three out of 53 tested normal tissues. This study used a new BeadChip combined with gene expression data in PCa to identify novel differentially methylated CpG sites located within genes. The newly identified differentially methylated genes may be used as biomarkers for PCa diagnosis.
许多在前列腺癌(PCa)中差异甲基化的基因已经被鉴定出来,主要使用基于候选基因的检测方法。最近,已经有几个关于 PCa 的全基因组 DNA 甲基化谱被报道,然而,这些方法在观察 PCa 中全基因组 DNA 甲基化改变的能力方面都存在弱点。我们假设在 PCa 中仍然存在未被识别的异常 DNA 甲基化,这些甲基化可能可以通过更高分辨率的检测方法来识别。我们使用新开发的 Illumina HumanMethylation450 BeadChip 在 PCa(n=19)和相邻正常组织(n=4)中进行了检测,并将这些数据与基因表达数据相结合,以鉴定在 PCa 发生和进展中可能具有功能后果的新的 DNA 甲基化。我们还在一个独立的数据集中证实了我们的甲基化结果。在另外的 56 个 PCa 样本和 55 个相邻正常组织中验证了两个异常 DNA 甲基化基因。共有 28735 个 CpG 位点在 DNA 甲基化上显示出显著差异(FDR 调整的 P<0.05),定义为 PCa 和正常样本之间的平均甲基化差异至少为 20%。此外,共有 122 个基因在其启动子区域有一个以上的差异甲基化 CpG 位点,并且基因表达模式与 DNA 甲基化变化的方向相反(例如,随着甲基化的增加表达减少,反之亦然)。通过亚硫酸氢盐测序证实了 AOX1 和 SPON2 两个基因的异常 DNA 甲基化,大多数相应的 CpG 位点在肿瘤样本和正常组织之间显示出显著差异。AOX1 启动子区域在 54 个测试的 PCa 样本中的 92.6%显示出过度甲基化,而在 53 个测试的正常组织中只有 3 个显示出过度甲基化。本研究使用新的 BeadChip 结合 PCa 中的基因表达数据,鉴定了位于基因内的新的差异甲基化 CpG 位点。新鉴定的差异甲基化基因可以用作 PCa 诊断的生物标志物。