Department of Statistics, Purdue University, 250 N. University Street, West Lafayette, Indiana, USA.
BMC Bioinformatics. 2012;13 Suppl 16(Suppl 16):S1. doi: 10.1186/1471-2105-13-S16-S1. Epub 2012 Nov 5.
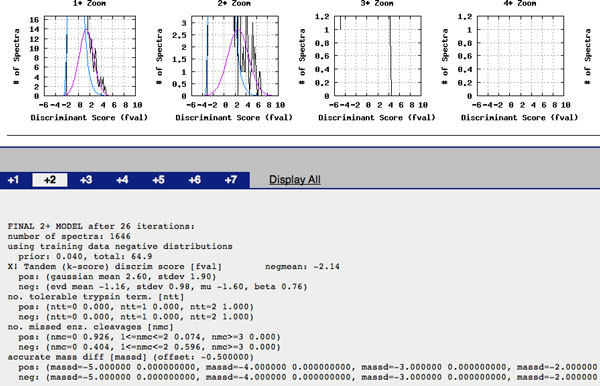
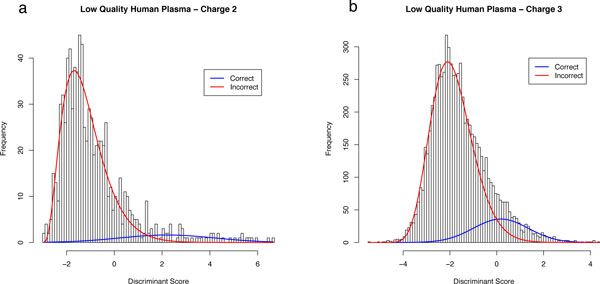
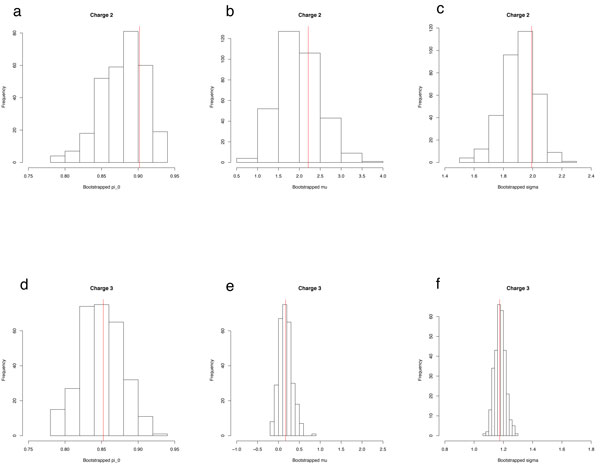
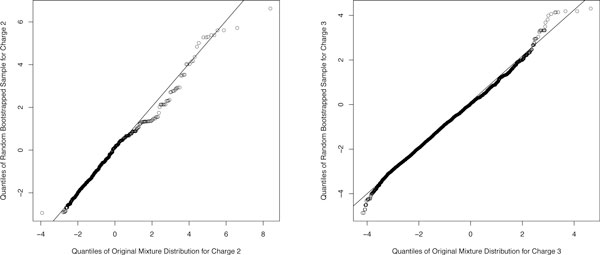
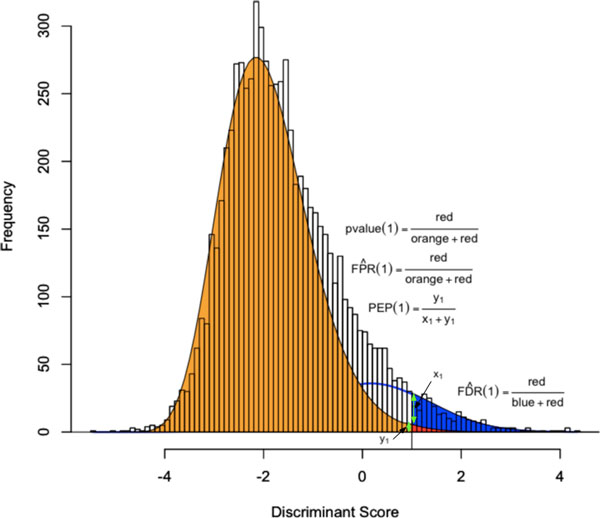
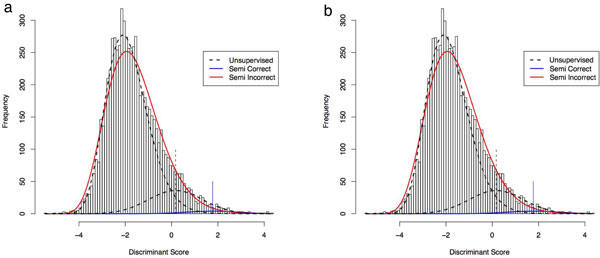
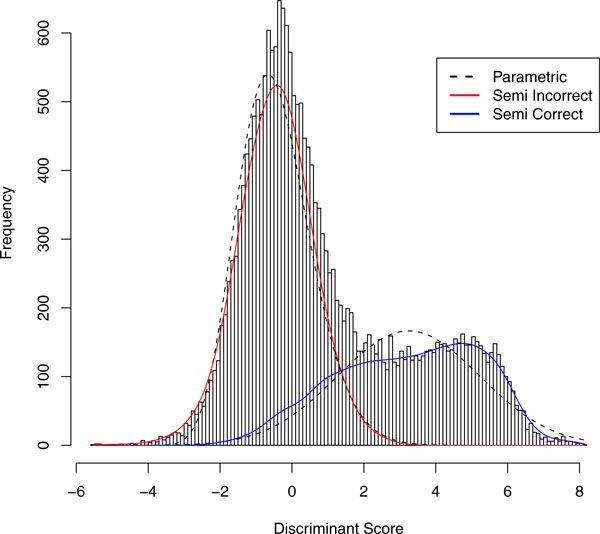
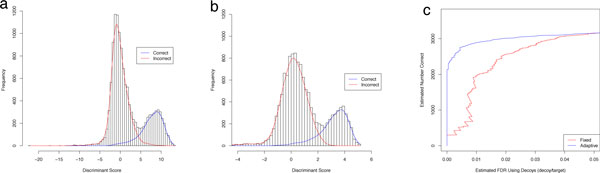
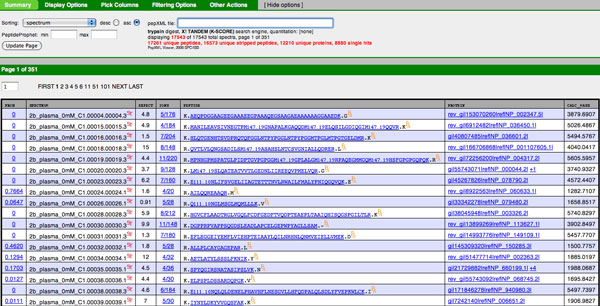
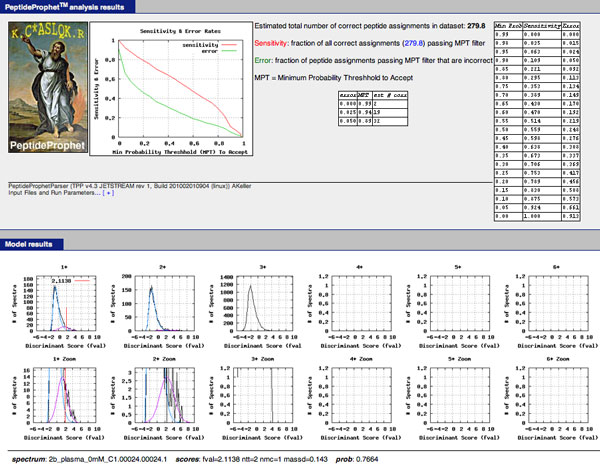
PeptideProphet is a post-processing algorithm designed to evaluate the confidence in identifications of MS/MS spectra returned by a database search. In this manuscript we describe the "what and how" of PeptideProphet in a manner aimed at statisticians and life scientists who would like to gain a more in-depth understanding of the underlying statistical modeling. The theory and rationale behind the mixture-modeling approach taken by PeptideProphet is discussed from a statistical model-building perspective followed by a description of how a model can be used to express confidence in the identification of individual peptides or sets of peptides. We also demonstrate how to evaluate the quality of model fit and select an appropriate model from several available alternatives. We illustrate the use of PeptideProphet in association with the Trans-Proteomic Pipeline, a free suite of software used for protein identification.
PeptideProphet 是一种后处理算法,旨在评估通过数据库搜索返回的 MS/MS 光谱鉴定的置信度。在本文中,我们以一种旨在让希望更深入了解基础统计建模的统计学家和生命科学家能够理解的方式,描述了 PeptideProphet 的“是什么”和“怎么做”。从统计模型构建的角度讨论了 PeptideProphet 采用的混合模型方法的理论和基本原理,然后描述了如何使用模型来表达对单个肽或肽集合鉴定的置信度。我们还展示了如何评估模型拟合的质量并从几个可用的替代方案中选择合适的模型。我们说明了如何将 PeptideProphet 与 Trans-Proteomic Pipeline 一起使用,Trans-Proteomic Pipeline 是一套免费的用于蛋白质鉴定的软件。