Institute of Biomaterials and Biomedical Engineering, University of Toronto, Toronto, Ontario, Canada.
PLoS Comput Biol. 2012;8(12):e1002838. doi: 10.1371/journal.pcbi.1002838. Epub 2012 Dec 20.
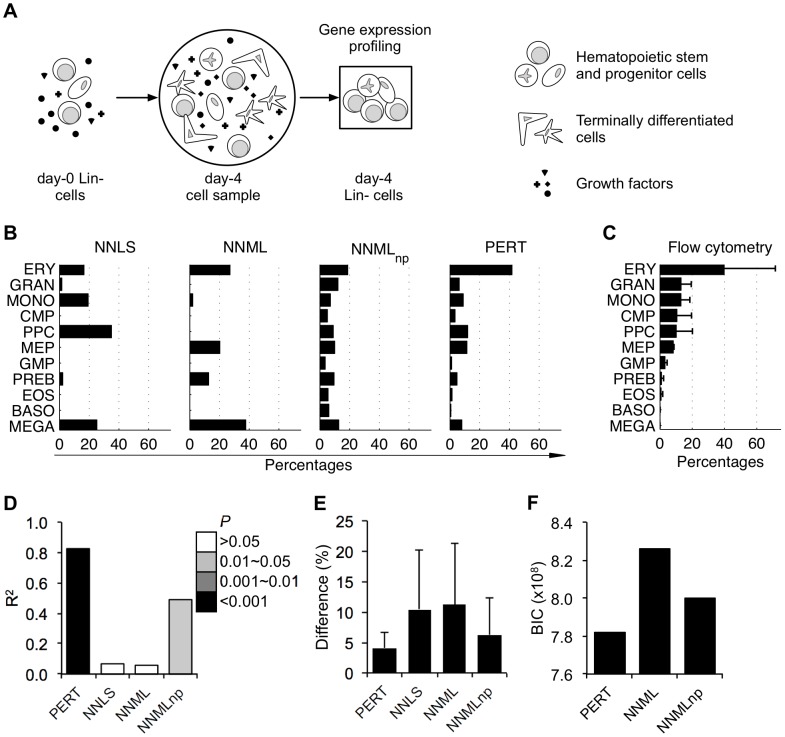
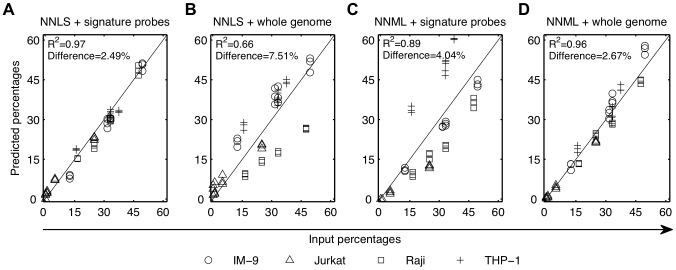
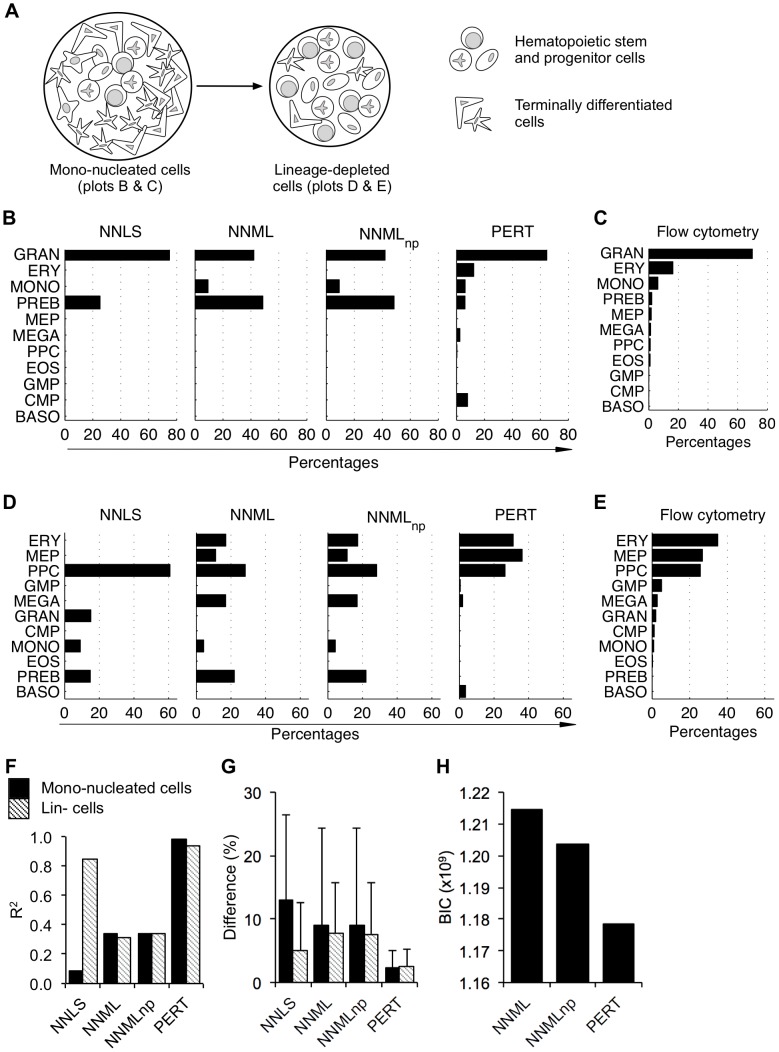
The cellular composition of heterogeneous samples can be predicted using an expression deconvolution algorithm to decompose their gene expression profiles based on pre-defined, reference gene expression profiles of the constituent populations in these samples. However, the expression profiles of the actual constituent populations are often perturbed from those of the reference profiles due to gene expression changes in cells associated with microenvironmental or developmental effects. Existing deconvolution algorithms do not account for these changes and give incorrect results when benchmarked against those measured by well-established flow cytometry, even after batch correction was applied. We introduce PERT, a new probabilistic expression deconvolution method that detects and accounts for a shared, multiplicative perturbation in the reference profiles when performing expression deconvolution. We applied PERT and three other state-of-the-art expression deconvolution methods to predict cell frequencies within heterogeneous human blood samples that were collected under several conditions (uncultured mono-nucleated and lineage-depleted cells, and culture-derived lineage-depleted cells). Only PERT's predicted proportions of the constituent populations matched those assigned by flow cytometry. Genes associated with cell cycle processes were highly enriched among those with the largest predicted expression changes between the cultured and uncultured conditions. We anticipate that PERT will be widely applicable to expression deconvolution strategies that use profiles from reference populations that vary from the corresponding constituent populations in cellular state but not cellular phenotypic identity.
使用表达分解算法可以根据这些样本中组成群体的预定义参考基因表达谱,对异质样本的细胞组成进行预测,从而分解它们的基因表达谱。然而,由于与微环境或发育效应相关的细胞中基因表达的变化,实际组成群体的表达谱往往与参考谱存在偏差。现有的分解算法并没有考虑到这些变化,即使在应用批量校正后,与通过成熟的流式细胞术测量的结果进行基准测试时,也会给出错误的结果。我们引入了 PERT,这是一种新的概率表达分解方法,在进行表达分解时,它可以检测并解释参考谱中存在的共享、乘法扰动。我们将 PERT 和其他三种最先进的表达分解方法应用于预测在几种条件下(未培养的单核细胞和谱系耗竭细胞,以及培养衍生的谱系耗竭细胞)收集的异质人类血液样本中的细胞频率。只有 PERT 预测的组成群体的比例与流式细胞术分配的比例相匹配。在培养和未培养条件之间,细胞周期过程相关的基因在预测表达变化最大的基因中高度富集。我们预计 PERT 将广泛适用于表达分解策略,这些策略使用来自参考群体的谱,这些参考群体在细胞状态上与相应的组成群体不同,但在细胞表型身份上没有差异。