Biomarker Development, Novartis Institutes for BioMedical Research, Cambridge, Massachusetts, United States of America.
PLoS One. 2011;6(11):e27156. doi: 10.1371/journal.pone.0027156. Epub 2011 Nov 16.
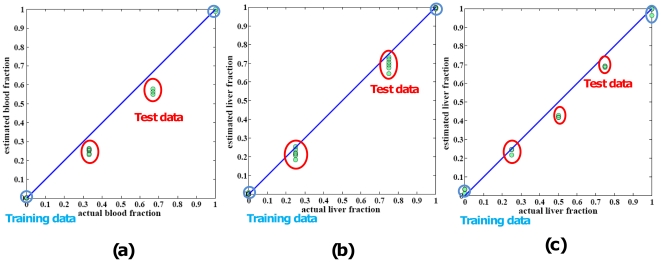
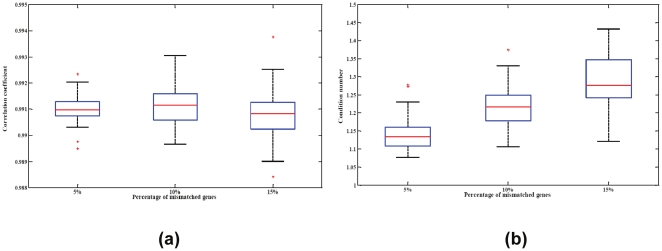
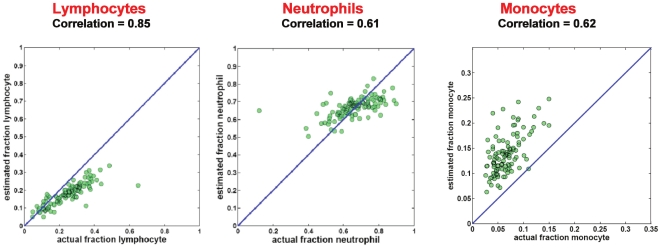
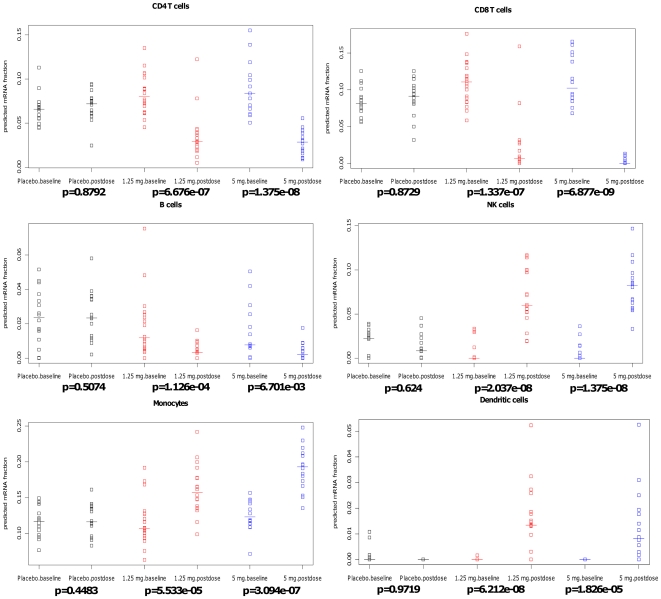
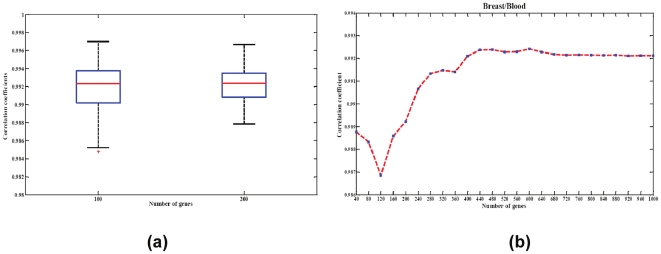
Large-scale molecular profiling technologies have assisted the identification of disease biomarkers and facilitated the basic understanding of cellular processes. However, samples collected from human subjects in clinical trials possess a level of complexity, arising from multiple cell types, that can obfuscate the analysis of data derived from them. Failure to identify, quantify, and incorporate sources of heterogeneity into an analysis can have widespread and detrimental effects on subsequent statistical studies.We describe an approach that builds upon a linear latent variable model, in which expression levels from mixed cell populations are modeled as the weighted average of expression from different cell types. We solve these equations using quadratic programming, which efficiently identifies the globally optimal solution while preserving non-negativity of the fraction of the cells. We applied our method to various existing platforms to estimate proportions of different pure cell or tissue types and gene expression profilings of distinct phenotypes, with a focus on complex samples collected in clinical trials. We tested our methods on several well controlled benchmark data sets with known mixing fractions of pure cell or tissue types and mRNA expression profiling data from samples collected in a clinical trial. Accurate agreement between predicted and actual mixing fractions was observed. In addition, our method was able to predict mixing fractions for more than ten species of circulating cells and to provide accurate estimates for relatively rare cell types (<10% total population). Furthermore, accurate changes in leukocyte trafficking associated with Fingolomid (FTY720) treatment were identified that were consistent with previous results generated by both cell counts and flow cytometry. These data suggest that our method can solve one of the open questions regarding the analysis of complex transcriptional data: namely, how to identify the optimal mixing fractions in a given experiment.
大规模分子分析技术有助于鉴定疾病生物标志物,并促进对细胞过程的基本理解。然而,临床试验中从人体样本中收集的样本具有一定程度的复杂性,来自多种细胞类型,这可能会使对从中获得的数据的分析变得复杂。未能识别、量化并将异质性来源纳入分析中会对随后的统计研究产生广泛而不利的影响。我们描述了一种基于线性潜在变量模型的方法,其中混合细胞群体的表达水平被建模为不同细胞类型表达的加权平均值。我们使用二次规划来求解这些方程,该方法有效地确定了全局最优解,同时保持细胞分数的非负性。我们将我们的方法应用于各种现有的平台,以估计不同纯细胞或组织类型的比例以及不同表型的基因表达谱,重点是在临床试验中收集的复杂样本。我们在几个具有已知纯细胞或组织类型混合分数的受控基准数据集和临床试验中收集的样本的 mRNA 表达谱数据上测试了我们的方法。观察到预测和实际混合分数之间的准确一致性。此外,我们的方法能够预测超过十种循环细胞的混合分数,并为相对罕见的细胞类型(<10%总群体)提供准确的估计。此外,还确定了与 Fingolomid(FTY720)治疗相关的白细胞迁移的准确变化,这与细胞计数和流式细胞术生成的先前结果一致。这些数据表明,我们的方法可以解决分析复杂转录数据的一个悬而未决的问题:即如何在给定实验中确定最佳混合分数。