Department of Neuroscience, Rosalind Franklin University/The Chicago Medical School, North Chicago, IL, USA.
PLoS One. 2012;7(12):e52056. doi: 10.1371/journal.pone.0052056. Epub 2012 Dec 21.
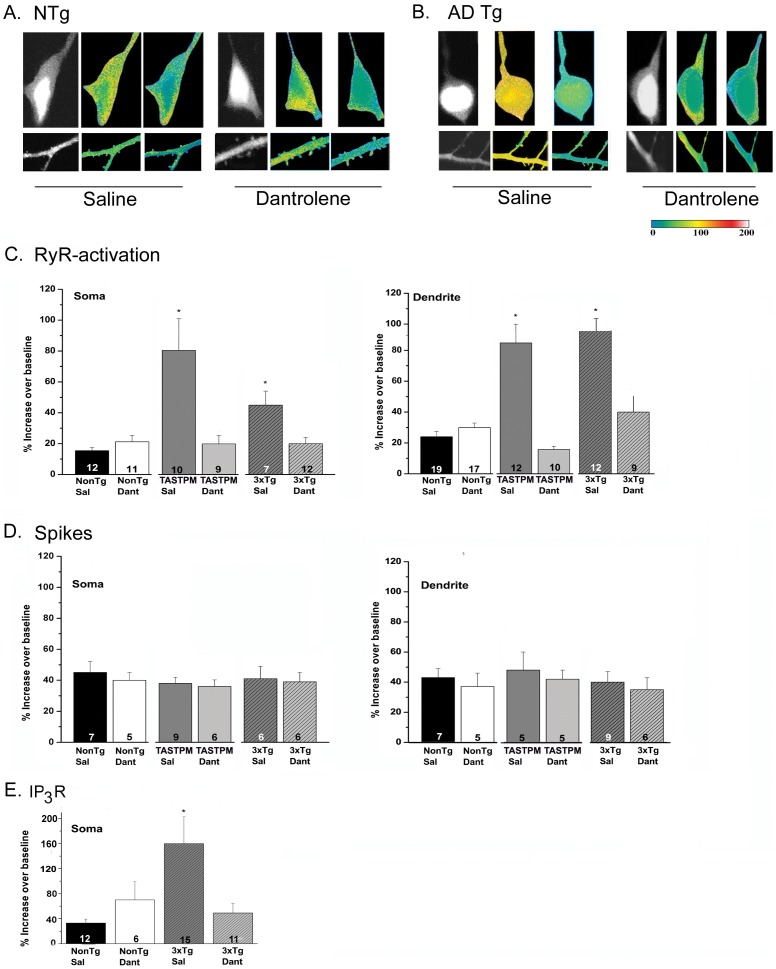
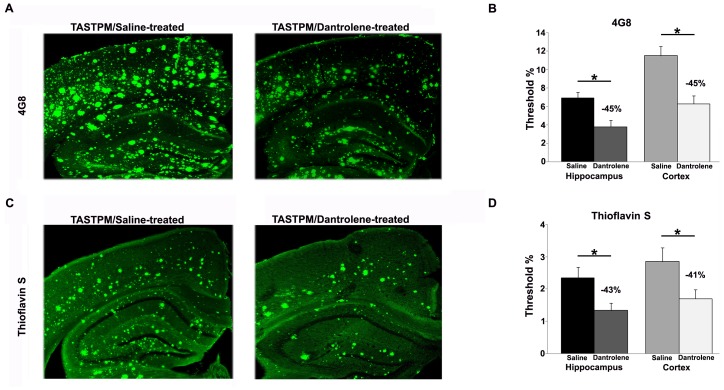
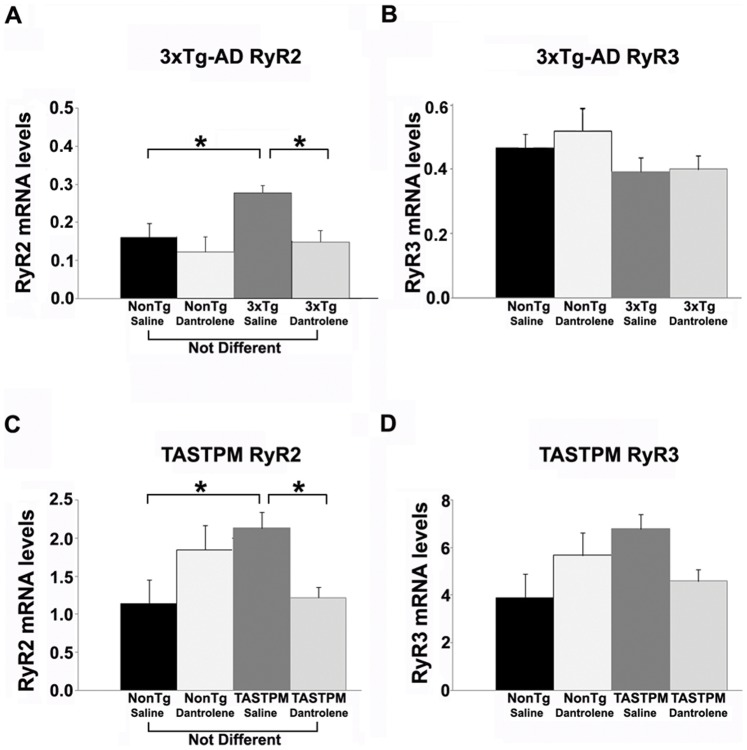
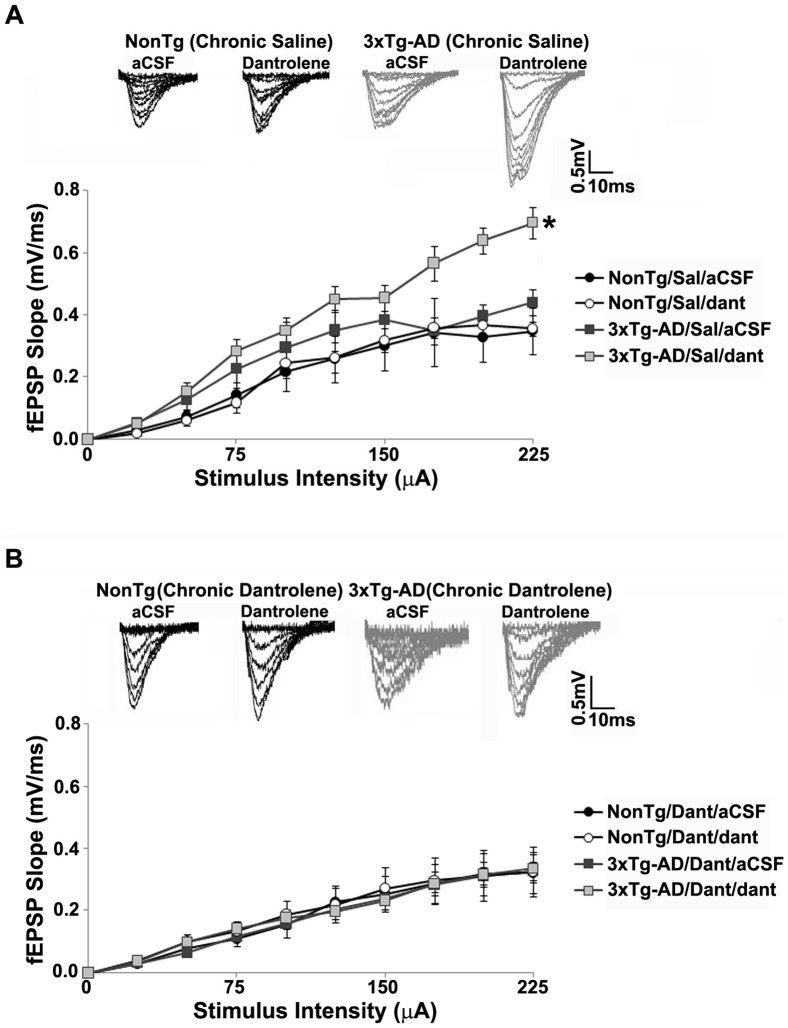
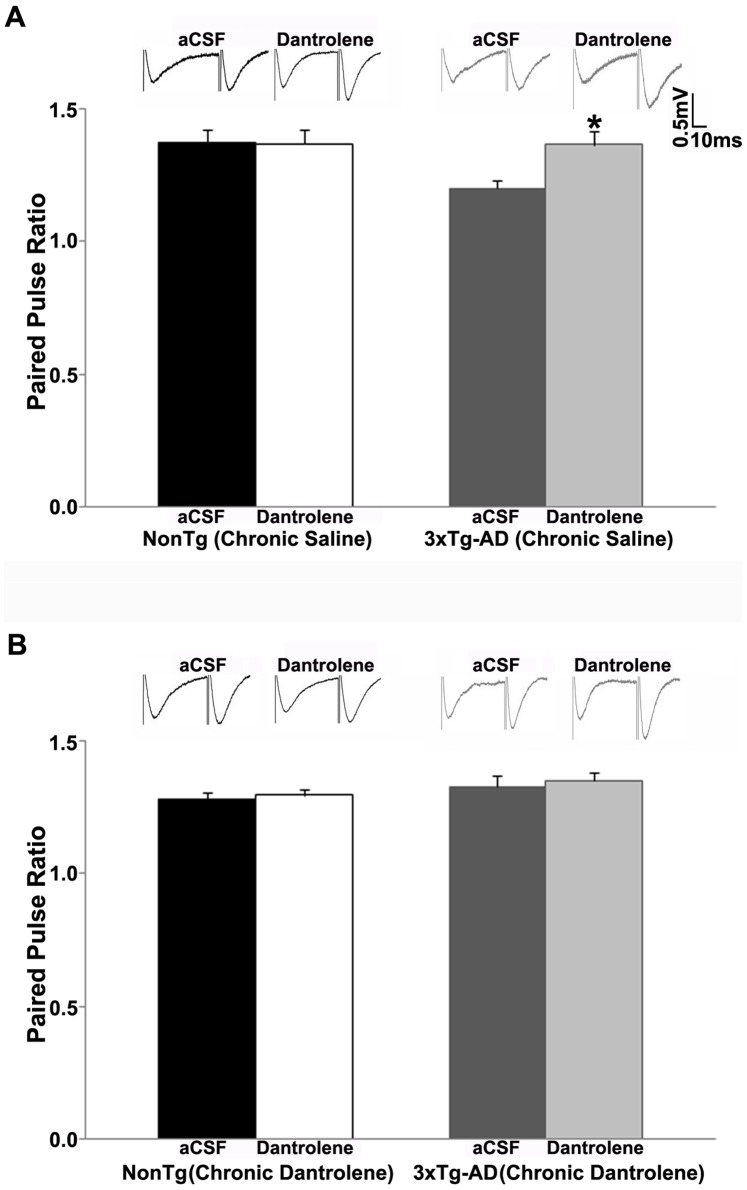
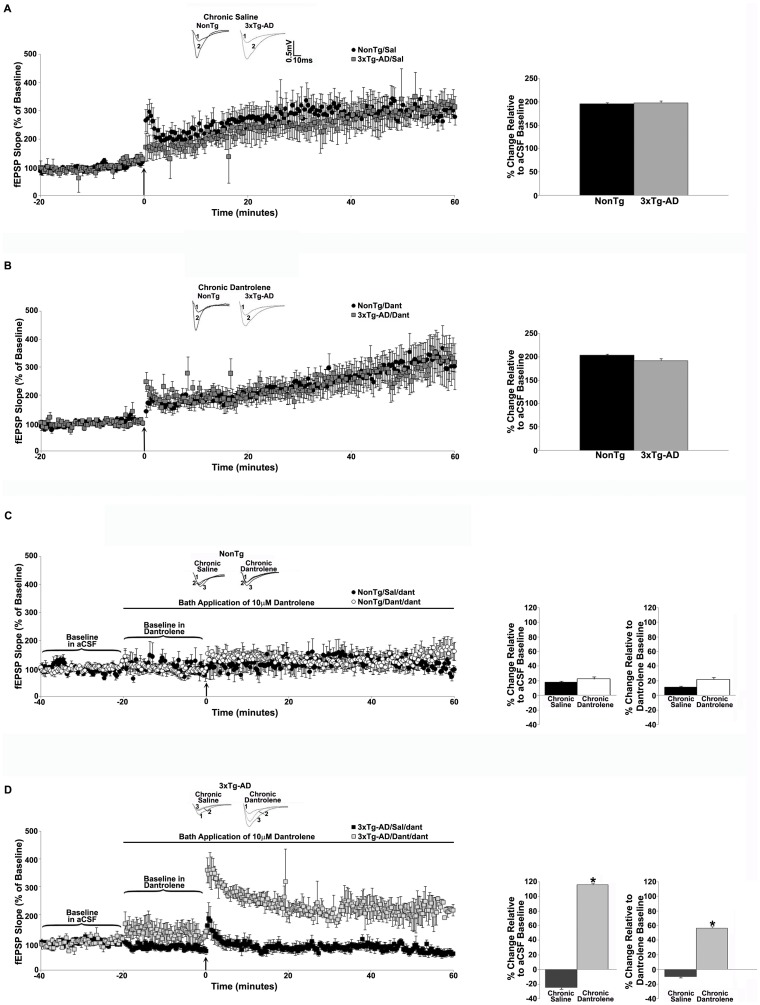
Alzheimer's disease (AD) is a devastating neurodegenerative condition with no known cure. While current therapies target late-stage amyloid formation and cholinergic tone, to date, these strategies have proven ineffective at preventing disease progression. The reasons for this may be varied, and could reflect late intervention, or, that earlier pathogenic mechanisms have been overlooked and permitted to accelerate the disease process. One such example would include synaptic pathology, the disease component strongly associated with cognitive impairment. Dysregulated Ca(2+) homeostasis may be one of the critical factors driving synaptic dysfunction. One of the earliest pathophysiological indicators in mutant presenilin (PS) AD mice is increased intracellular Ca(2+) signaling, predominantly through the ER-localized inositol triphosphate (IP(3)) and ryanodine receptors (RyR). In particular, the RyR-mediated Ca(2+) upregulation within synaptic compartments is associated with altered synaptic homeostasis and network depression at early (presymptomatic) AD stages. Here, we offer an alternative approach to AD therapeutics by stabilizing early pathogenic mechanisms associated with synaptic abnormalities. We targeted the RyR as a means to prevent disease progression, and sub-chronically treated AD mouse models (4-weeks) with a novel formulation of the RyR inhibitor, dantrolene. Using 2-photon Ca(2+) imaging and patch clamp recordings, we demonstrate that dantrolene treatment fully normalizes ER Ca(2+) signaling within somatic and dendritic compartments in early and later-stage AD mice in hippocampal slices. Additionally, the elevated RyR2 levels in AD mice are restored to control levels with dantrolene treatment, as are synaptic transmission and synaptic plasticity. Aβ deposition within the cortex and hippocampus is also reduced in dantrolene-treated AD mice. In this study, we highlight the pivotal role of Ca(2+) aberrations in AD, and propose a novel strategy to preserve synaptic function, and thereby cognitive function, in early AD patients.
阿尔茨海默病(AD)是一种破坏性的神经退行性疾病,目前尚无已知的治愈方法。虽然目前的治疗方法针对晚期淀粉样蛋白形成和胆碱能张力,但迄今为止,这些策略在预防疾病进展方面都证明是无效的。原因可能是多种多样的,可能反映出干预时间较晚,或者早期的致病机制被忽视,从而加速了疾病的进程。一个这样的例子是突触病理学,与认知障碍强烈相关的疾病成分。钙稳态失调可能是导致突触功能障碍的关键因素之一。突变早老素(PS)AD 小鼠中最早的病理生理学指标之一是细胞内 Ca(2+)信号的增加,主要通过内质网定位的三磷酸肌醇(IP(3))和兰尼碱受体(RyR)。特别是,突触间隙中 RyR 介导的 Ca(2+)上调与早期(无症状前)AD 阶段的突触稳态改变和网络抑制有关。在这里,我们通过稳定与突触异常相关的早期致病机制,提供了一种治疗 AD 的替代方法。我们将 RyR 作为一种预防疾病进展的手段,并用一种新型的 RyR 抑制剂,丹曲林钠,对 AD 小鼠模型进行了亚慢性治疗(4 周)。通过双光子 Ca(2+)成像和膜片钳记录,我们证明丹曲林钠治疗可使 AD 早期和晚期小鼠海马切片中体细胞和树突体区的内质网 Ca(2+)信号完全正常化。此外,AD 小鼠中升高的 RyR2 水平在丹曲林钠治疗后恢复到对照水平,突触传递和突触可塑性也是如此。丹曲林钠治疗还可减少 AD 小鼠皮质和海马内的 Aβ沉积。在这项研究中,我们强调了 Ca(2+)异常在 AD 中的关键作用,并提出了一种新的策略来保护早期 AD 患者的突触功能,从而保护认知功能。