Centre de Recherche en Nutrition Humaine Auvergne, Clermont Université, Université d'Auvergne, EA 4678, Conception, Ingénierie et Développement de l'Aliment et du Médicament, BP 10448, Clermont-Ferrand 63000, France.
DNA Res. 2013 Apr;20(2):185-96. doi: 10.1093/dnares/dst001. Epub 2013 Jan 30.
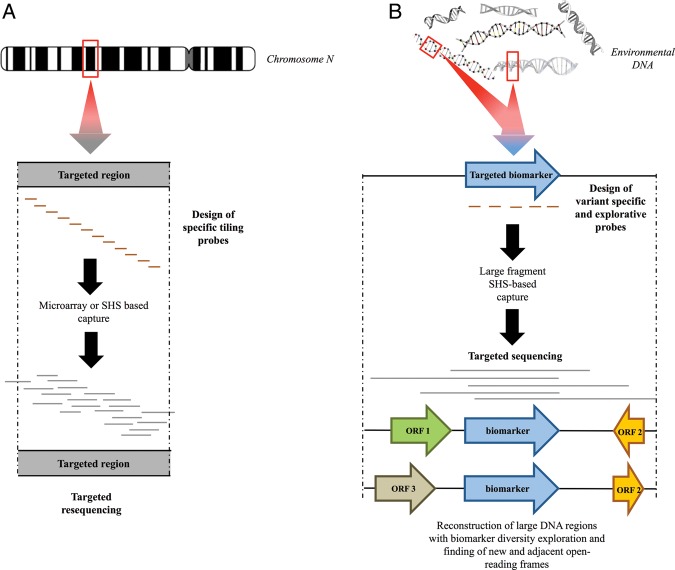
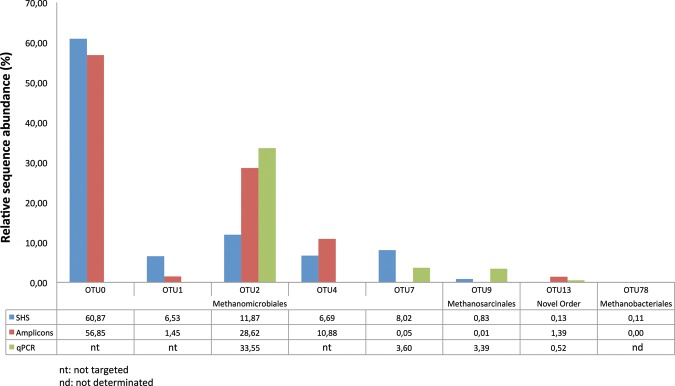
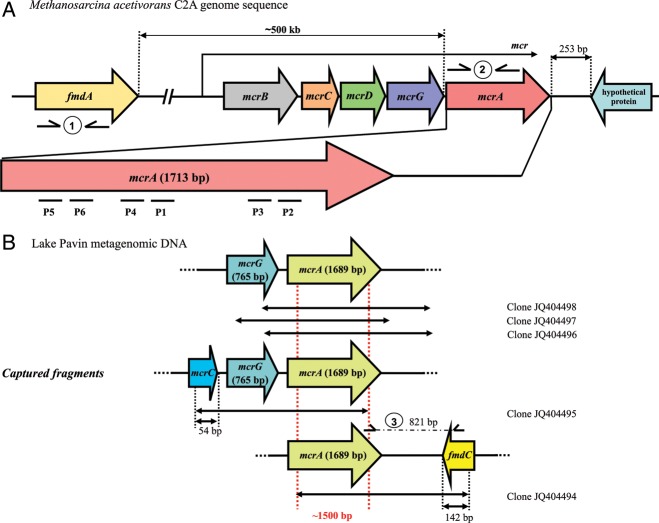
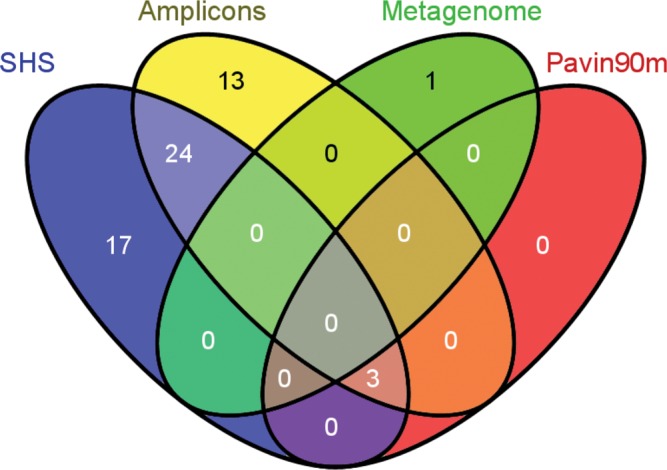
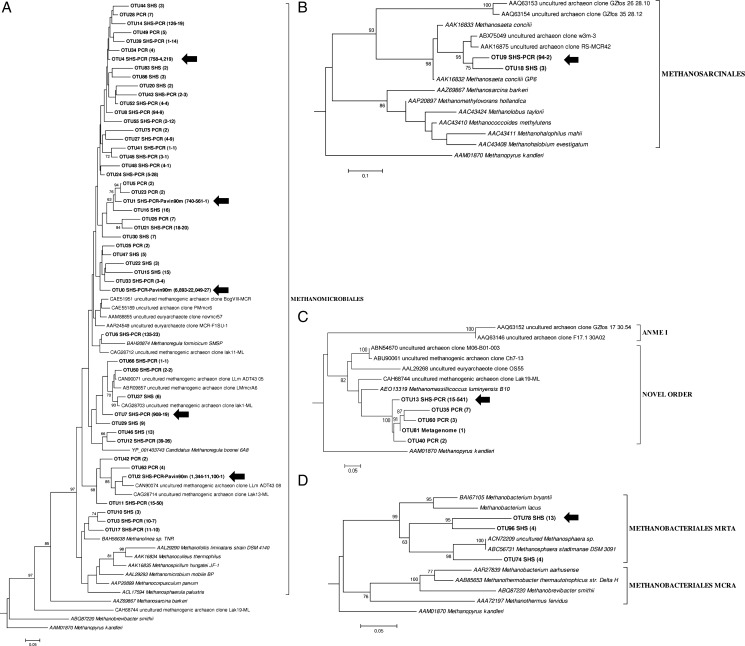
Next-generation sequencing (NGS) allows faster acquisition of metagenomic data, but complete exploration of complex ecosystems is hindered by the extraordinary diversity of microorganisms. To reduce the environmental complexity, we created an innovative solution hybrid selection (SHS) method that is combined with NGS to characterize large DNA fragments harbouring biomarkers of interest. The quality of enrichment was evaluated after fragments containing the methyl coenzyme M reductase subunit A gene (mcrA), the biomarker of methanogenesis, were captured from a Methanosarcina strain and a metagenomic sample from a meromictic lake. The methanogen diversity was compared with direct metagenome and mcrA-based amplicon pyrosequencing strategies. The SHS approach resulted in the capture of DNA fragments up to 2.5 kb with an enrichment efficiency between 41 and 100%, depending on the sample complexity. Compared with direct metagenome and amplicons sequencing, SHS detected broader mcrA diversity, and it allowed efficient sampling of the rare biosphere and unknown sequences. In contrast to amplicon-based strategies, SHS is less biased and GC independent, and it recovered complete biomarker sequences in addition to conserved regions. Because this method can also isolate the regions flanking the target sequences, it could facilitate operon reconstructions.
下一代测序(NGS)可以更快地获取宏基因组数据,但由于微生物的非凡多样性,对复杂生态系统的完全探索受到阻碍。为了降低环境复杂性,我们创建了一种创新的混合选择(SHS)方法,该方法与 NGS 相结合,用于描述含有感兴趣生物标志物的大 DNA 片段。从 Methanosarcina 菌株和分层湖的宏基因组样本中捕获含有甲基辅酶 M 还原酶亚基 A 基因(mcrA)的片段后,评估了富集的质量,mcrA 是产甲烷生物的生物标志物。将甲烷生物多样性与直接宏基因组和基于 mcrA 的扩增子焦磷酸测序策略进行了比较。SHS 方法可捕获长达 2.5 kb 的 DNA 片段,富集效率取决于样品复杂性,在 41%到 100%之间。与直接宏基因组和扩增子测序相比,SHS 检测到更广泛的 mcrA 多样性,并能够有效地对稀有生物圈和未知序列进行采样。与基于扩增子的策略相比,SHS 的偏差较小,不依赖 GC,并且除了保守区域外,还可以回收完整的生物标志物序列。由于该方法还可以分离目标序列侧翼的区域,因此它可以促进操纵子的重建。