Department of Medicine, University of Alberta Edmonton, AB, Canada.
Front Oncol. 2013 Mar 7;3:38. doi: 10.3389/fonc.2013.00038. eCollection 2013.
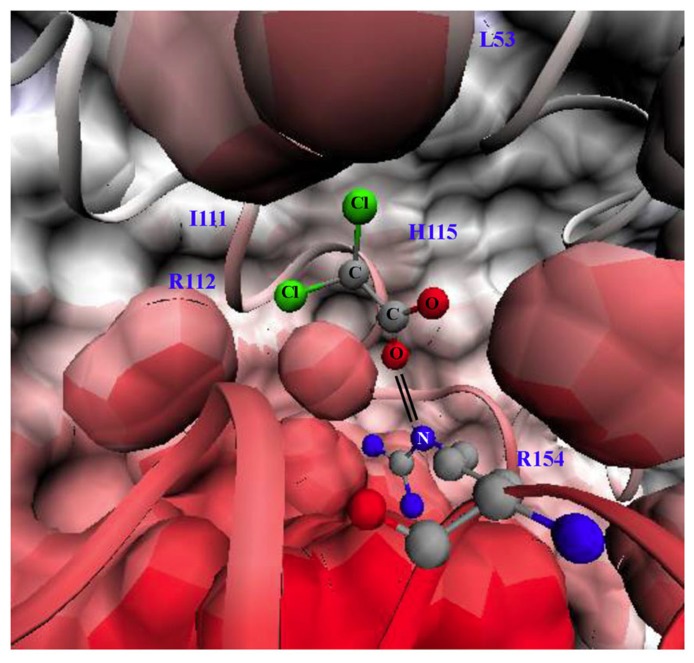
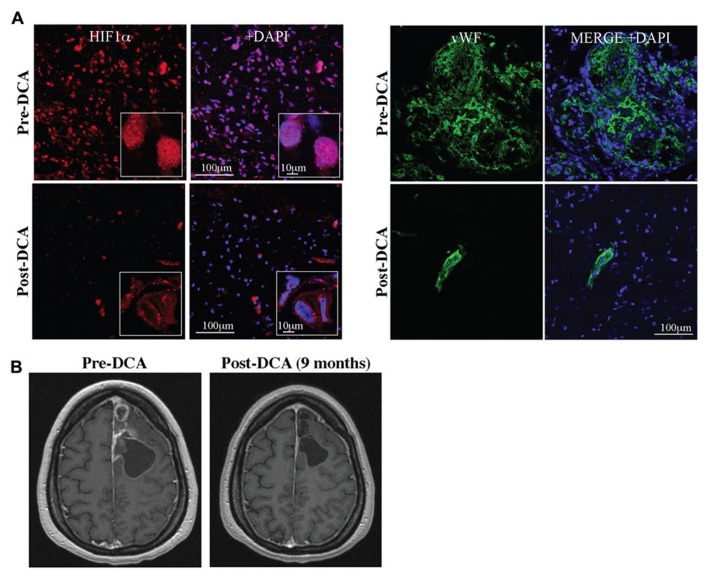
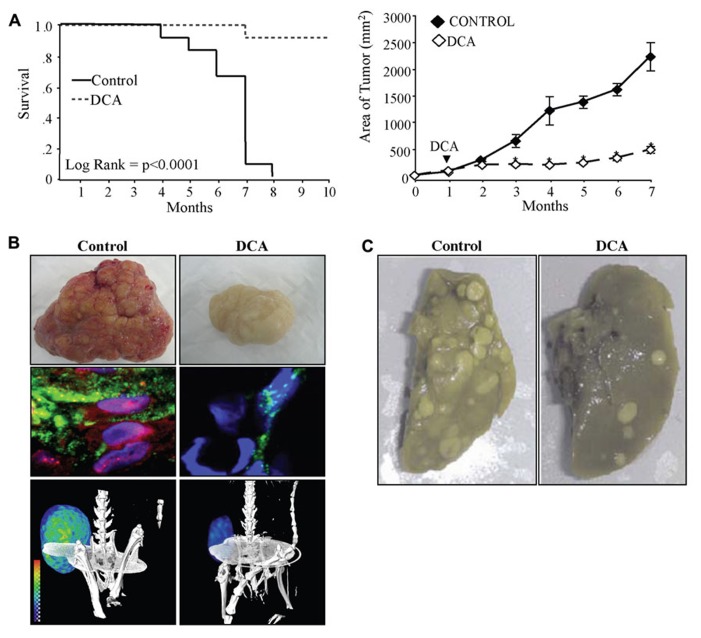
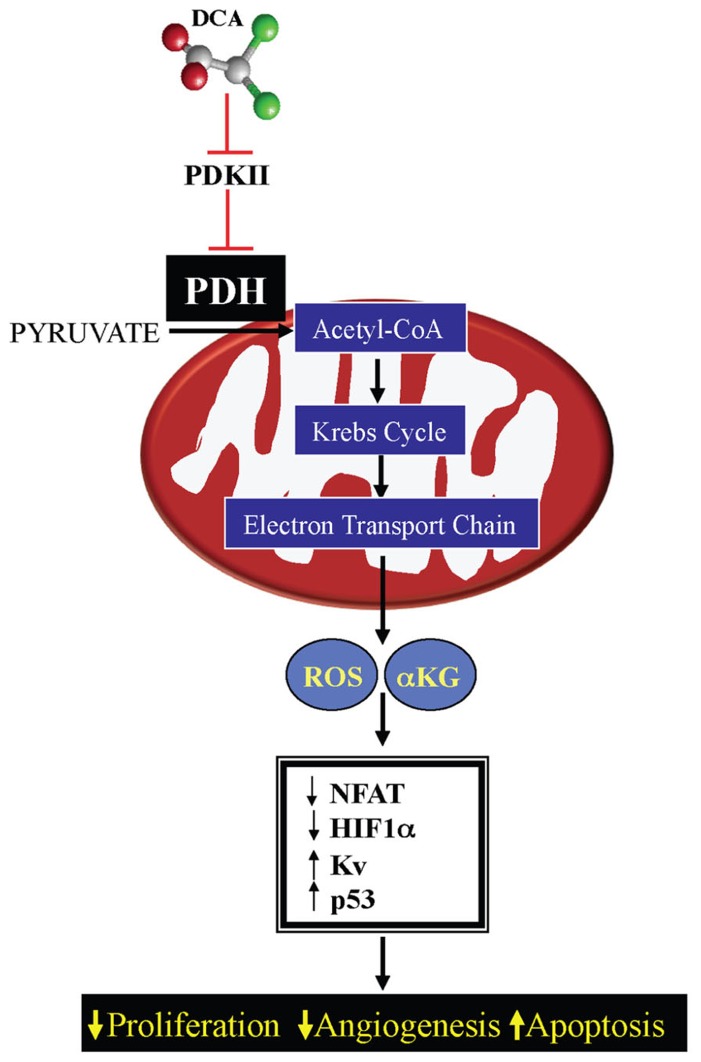
Current drug development in oncology is non-selective as it typically focuses on pathways essential for the survival of all dividing cells. The unique metabolic profile of cancer, which is characterized by increased glycolysis and suppressed mitochondrial glucose oxidation (GO) provides cancer cells with a proliferative advantage, conducive with apoptosis resistance and even increased angiogenesis. Recent evidence suggests that targeting the cancer-specific metabolic and mitochondrial remodeling may offer selectivity in cancer treatment. Pyruvate dehydrogenase kinase (PDK) is a mitochondrial enzyme that is activated in a variety of cancers and results in the selective inhibition of pyruvate dehydrogenase, a complex of enzymes that converts cytosolic pyruvate to mitochondrial acetyl-CoA, the substrate for the Krebs' cycle. Inhibition of PDK with either small interfering RNAs or the orphan drug dichloroacetate (DCA) shifts the metabolism of cancer cells from glycolysis to GO and reverses the suppression of mitochondria-dependent apoptosis. In addition, this therapeutic strategy increases the production of diffusible Krebs' cycle intermediates and mitochondria-derived reactive oxygen species, activating p53 or inhibiting pro-proliferative and pro-angiogenic transcription factors like nuclear factor of activated T cells and hypoxia-inducible factor 1α. These effects result in decreased tumor growth and angiogenesis in a variety of cancers with high selectivity. In a small but mechanistic clinical trial in patients with glioblastoma, a highly aggressive and vascular form of brain cancer, DCA decreased tumor angiogenesis and tumor growth, suggesting that metabolic-targeting therapies can be translated directly to patients. More recently, the M2 isoform of pyruvate kinase (PKM2), which is highly expressed in cancer, is associated with suppressed mitochondrial function. Similar to DCA, activation of PKM2 in many cancers results in increased mitochondrial function and decreased tumor growth. Therefore, reversing the mitochondrial suppression with metabolic-modulating drugs, like PDK inhibitors or PKM2 activators holds promise in the rapidly expanding field of metabolic oncology.
当前的肿瘤学药物研发是非选择性的,因为它通常侧重于所有分裂细胞生存所必需的途径。癌症独特的代谢特征是糖酵解增加而线粒体葡萄糖氧化(GO)受到抑制,这为癌细胞提供了增殖优势,有利于抗凋亡甚至增加血管生成。最近的证据表明,针对癌症特异性代谢和线粒体重塑可能为癌症治疗提供选择性。丙酮酸脱氢酶激酶(PDK)是一种线粒体酶,在多种癌症中被激活,导致丙酮酸脱氢酶的选择性抑制,丙酮酸脱氢酶是一种将细胞质丙酮酸转化为线粒体乙酰辅酶 A 的酶复合物,乙酰辅酶 A 是三羧酸循环的底物。使用小干扰 RNA 或孤儿药物二氯乙酸(DCA)抑制 PDK 会使癌细胞的代谢从糖酵解转变为 GO,并逆转线粒体依赖性细胞凋亡的抑制。此外,这种治疗策略增加了可扩散的三羧酸循环中间产物和线粒体来源的活性氧的产生,激活 p53 或抑制促增殖和促血管生成转录因子,如激活 T 细胞的核因子和缺氧诱导因子 1α。这些效应导致各种癌症的肿瘤生长和血管生成减少,具有高度选择性。在一项针对胶质母细胞瘤(一种高度侵袭性和血管性脑癌)患者的小型但具有机制性的临床试验中,DCA 降低了肿瘤血管生成和肿瘤生长,表明代谢靶向疗法可以直接应用于患者。最近,在癌症中高度表达的丙酮酸激酶(PKM2)的 M2 同工型与线粒体功能受抑制有关。与 DCA 类似,许多癌症中 PKM2 的激活导致线粒体功能增加和肿瘤生长减少。因此,使用代谢调节药物(如 PDK 抑制剂或 PKM2 激活剂)逆转线粒体抑制在代谢肿瘤学这一日益扩大的领域中具有广阔的前景。