Department of Genetics and Evolution, Institute of Genetics and Genomics in Geneva (iGE3), University of Geneva, Geneva, Switzerland.
BMC Genomics. 2013 Mar 25;14:204. doi: 10.1186/1471-2164-14-204.
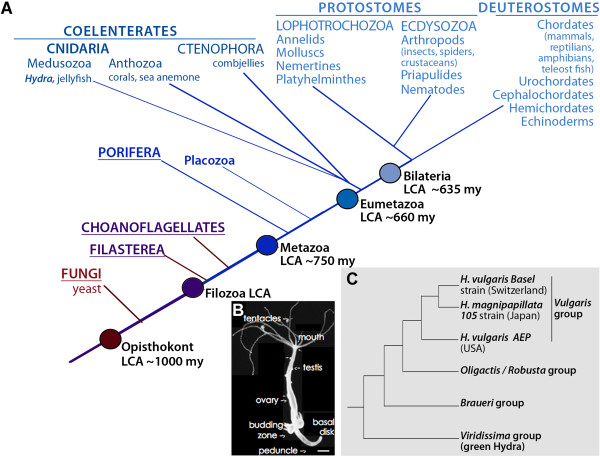
Evolutionary studies benefit from deep sequencing technologies that generate genomic and transcriptomic sequences from a variety of organisms. Genome sequencing and RNAseq have complementary strengths. In this study, we present the assembly of the most complete Hydra transcriptome to date along with a comparative analysis of the specific features of RNAseq and genome-predicted transcriptomes currently available in the freshwater hydrozoan Hydra vulgaris.
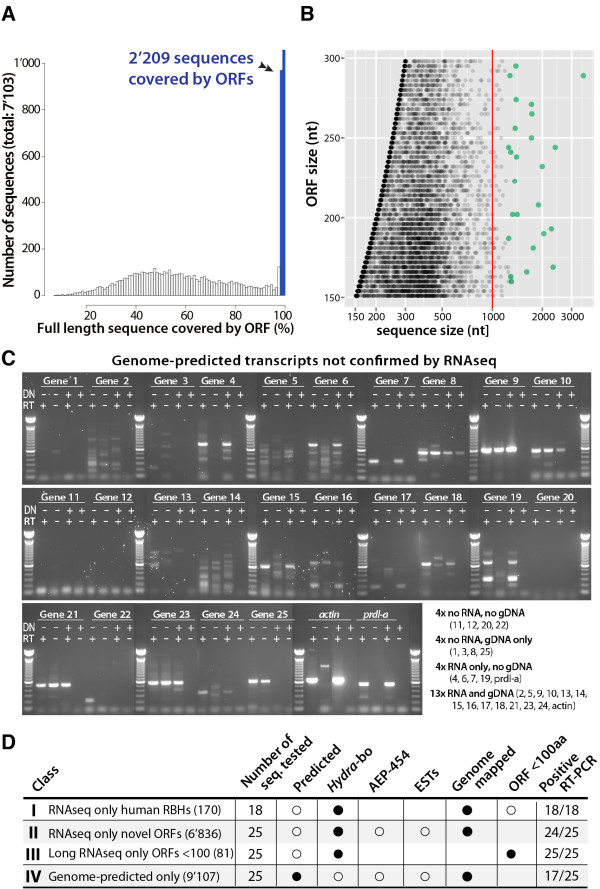
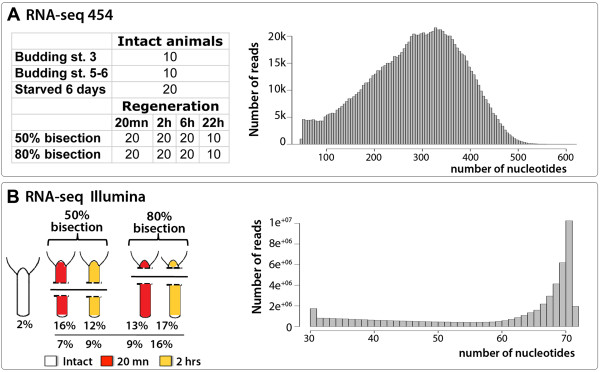
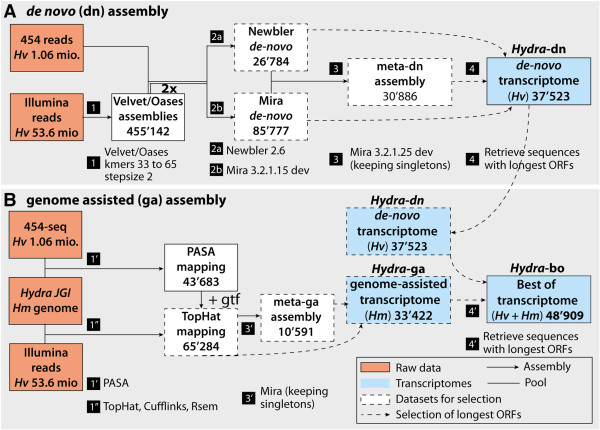
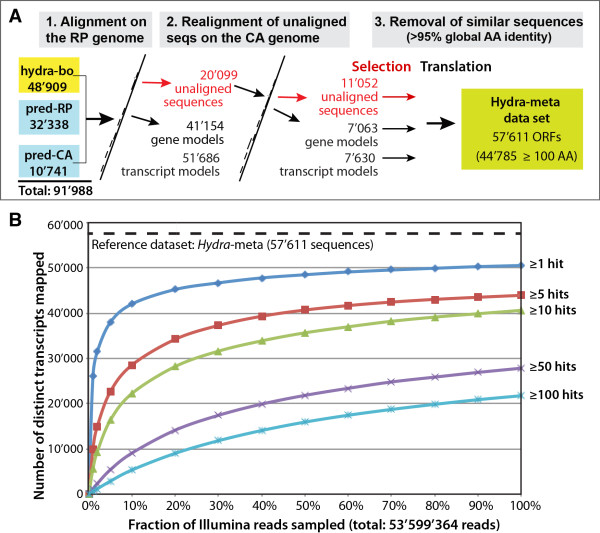
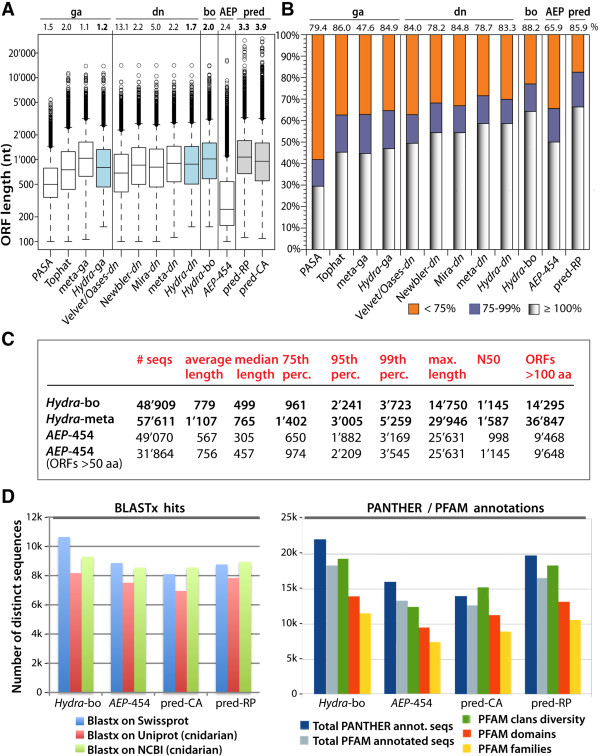
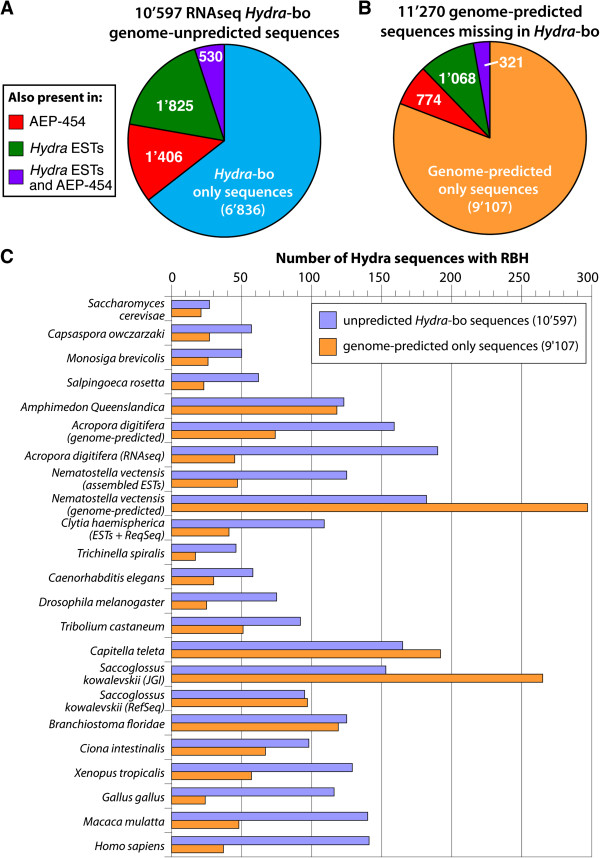
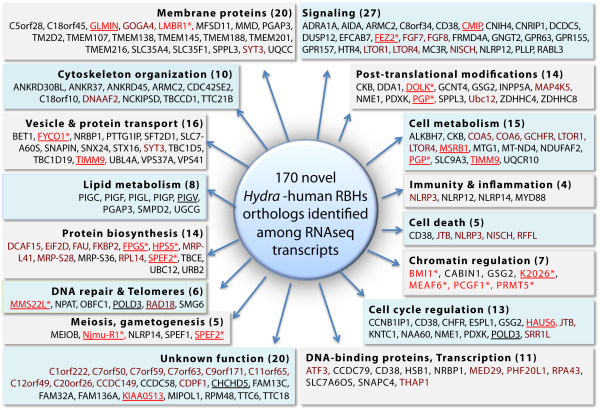
To produce an accurate and extensive Hydra transcriptome, we combined Illumina and 454 Titanium reads, giving the primacy to Illumina over 454 reads to correct homopolymer errors. This strategy yielded an RNAseq transcriptome that contains 48'909 unique sequences including splice variants, representing approximately 24'450 distinct genes. Comparative analysis to the available genome-predicted transcriptomes identified 10'597 novel Hydra transcripts that encode 529 evolutionarily-conserved proteins. The annotation of 170 human orthologs points to critical functions in protein biosynthesis, FGF and TOR signaling, vesicle transport, immunity, cell cycle regulation, cell death, mitochondrial metabolism, transcription and chromatin regulation. However, a majority of these novel transcripts encodes short ORFs, at least 767 of them corresponding to pseudogenes. This RNAseq transcriptome also lacks 11'270 predicted transcripts that correspond either to silent genes or to genes expressed below the detection level of this study.
We established a simple and powerful strategy to combine Illumina and 454 reads and we produced, with genome assistance, an extensive and accurate Hydra transcriptome. The comparative analysis of the RNAseq transcriptome with genome-predicted transcriptomes lead to the identification of large populations of novel as well as missing transcripts that might reflect Hydra-specific evolutionary events.
进化研究受益于深度测序技术,该技术可从各种生物体中生成基因组和转录组序列。基因组测序和 RNAseq 具有互补优势。在这项研究中,我们展示了迄今为止最完整的水螅转录组的组装,以及对淡水水螅属水螅现有的 RNAseq 和基于基因组预测的转录组的特定特征的比较分析。
为了生成准确和广泛的水螅转录组,我们结合了 Illumina 和 454 钛读取,优先使用 Illumina 而不是 454 读取来纠正同源多聚体错误。这种策略产生的 RNAseq 转录组包含 48909 个独特序列,包括剪接变体,代表大约 24450 个不同的基因。与现有基因组预测转录组的比较分析确定了 10597 个新的水螅转录本,它们编码 529 个进化保守的蛋白质。170 个人类同源物的注释指向蛋白质生物合成、FGF 和 TOR 信号、囊泡运输、免疫、细胞周期调控、细胞死亡、线粒体代谢、转录和染色质调控的关键功能。然而,这些新的转录本中的大多数编码短的 ORF,其中至少有 767 个对应于假基因。这个 RNAseq 转录组也缺少 11270 个预测的转录本,它们要么对应于沉默基因,要么对应于本研究检测水平以下表达的基因。
我们建立了一种简单而强大的策略,结合了 Illumina 和 454 读取,并在基因组的帮助下,生成了一个广泛而准确的水螅转录组。RNAseq 转录组与基于基因组预测的转录组的比较分析导致了大量新的和缺失的转录本的鉴定,这些转录本可能反映了水螅特有的进化事件。