Department of Computer Science, University of California-Irvine, CA, USA.
BMC Bioinformatics. 2013;14 Suppl 5(Suppl 5):S4. doi: 10.1186/1471-2105-14-S5-S4. Epub 2013 Apr 10.
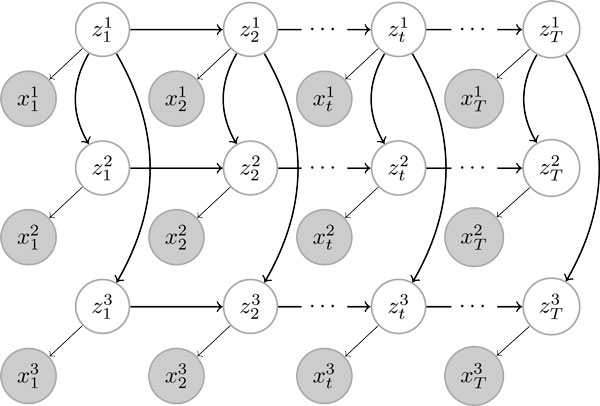
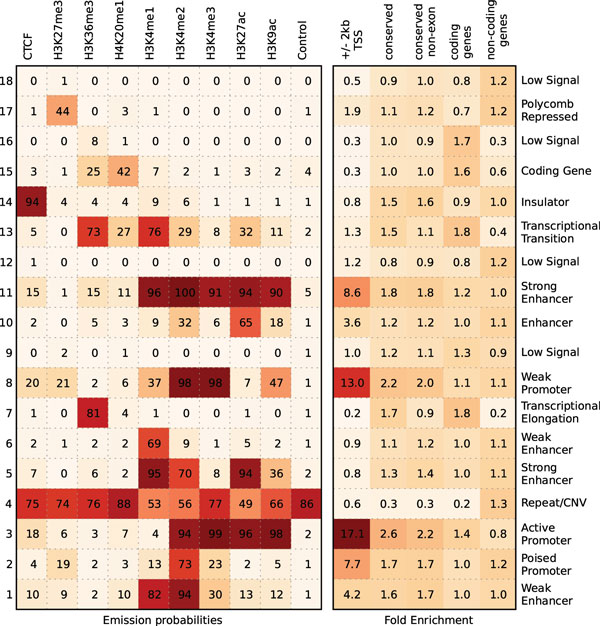
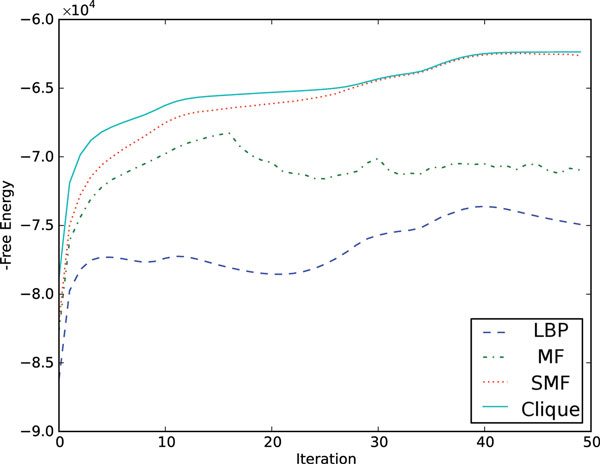
New biological techniques and technological advances in high-throughput sequencing are paving the way for systematic, comprehensive annotation of many genomes, allowing differences between cell types or between disease/normal tissues to be determined with unprecedented breadth. Epigenetic modifications have been shown to exhibit rich diversity between cell types, correlate tightly with cell-type specific gene expression, and changes in epigenetic modifications have been implicated in several diseases. Previous attempts to understand chromatin state have focused on identifying combinations of epigenetic modification, but in cases of multiple cell types, have not considered the lineage of the cells in question.We present a Bayesian network that uses epigenetic modifications to simultaneously model 1) chromatin mark combinations that give rise to different chromatin states and 2) propensities for transitions between chromatin states through differentiation or disease progression. We apply our model to a recent dataset of histone modifications, covering nine human cell types with nine epigenetic modifications measured for each. Since exact inference in this model is intractable for all the scale of the datasets, we develop several variational approximations and explore their accuracy. Our method exhibits several desirable features including improved accuracy of inferring chromatin states, improved handling of missing data, and linear scaling with dataset size. The source code for our model is available at http:// http://github.com/uci-cbcl/tree-hmm.
新的生物技术和高通量测序技术的进步为系统地、全面地注释许多基因组铺平了道路,使细胞类型之间或疾病/正常组织之间的差异能够以前所未有的广度来确定。已经表明,表观遗传修饰在细胞类型之间表现出丰富的多样性,与细胞类型特异性基因表达密切相关,并且表观遗传修饰的变化与几种疾病有关。以前尝试理解染色质状态的方法集中于识别表观遗传修饰的组合,但在涉及多种细胞类型的情况下,没有考虑到所讨论的细胞的谱系。我们提出了一个贝叶斯网络,该网络使用表观遗传修饰来同时建模 1)产生不同染色质状态的染色质标记组合,以及 2)通过分化或疾病进展在染色质状态之间进行转换的倾向。我们将我们的模型应用于最近的组蛋白修饰数据集,该数据集涵盖了九个人类细胞类型,每个细胞类型测量了九个表观遗传修饰。由于在所有数据集的规模上,这个模型的精确推断都是难以处理的,因此我们开发了几种变分近似,并探索了它们的准确性。我们的方法具有几个理想的特征,包括提高推断染色质状态的准确性,更好地处理缺失数据,以及与数据集大小的线性缩放。我们模型的源代码可在 http:// http://github.com/uci-cbcl/tree-hmm 上获得。