Department of Genetics and Biotechnology, St. Petersburg State University, St. Petersburg, Russia.
Evol Bioinform Online. 2013 Jul 11;9:263-73. doi: 10.4137/EBO.S12299. Print 2013.
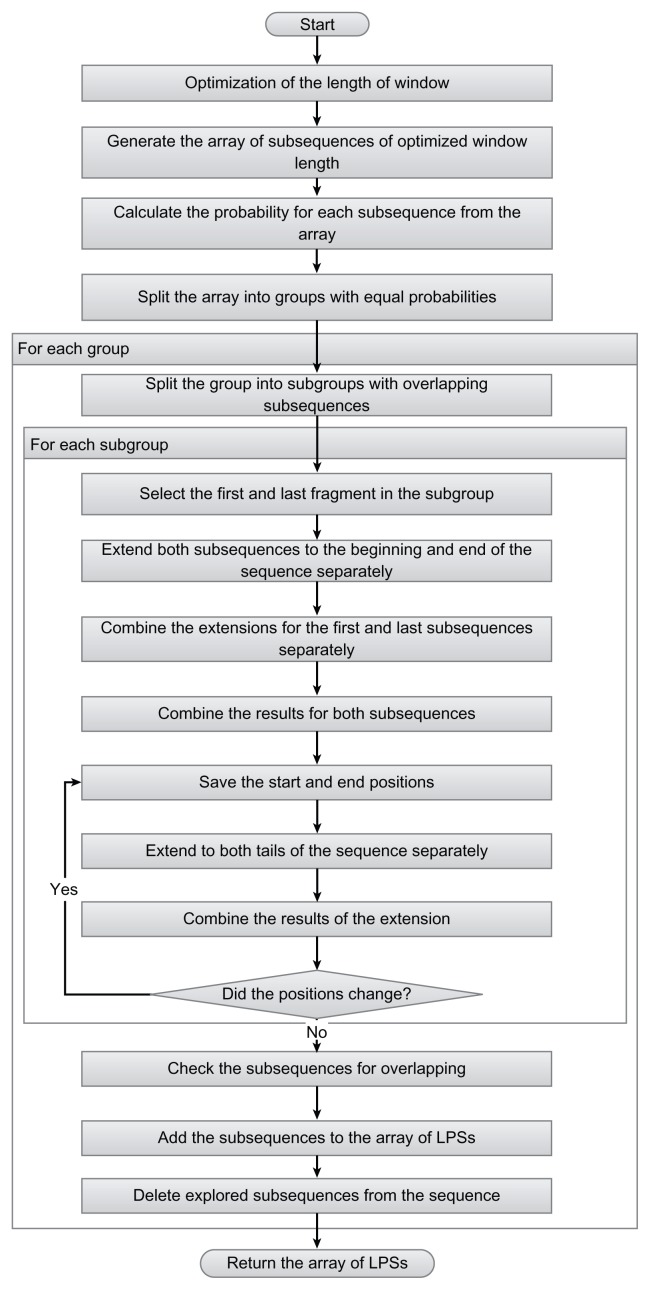
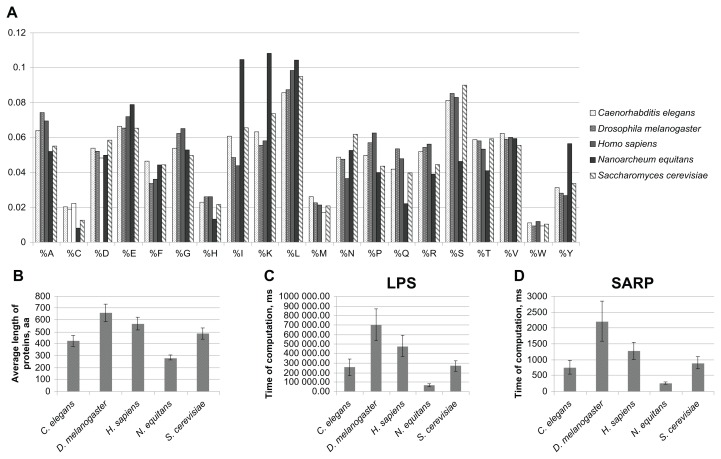
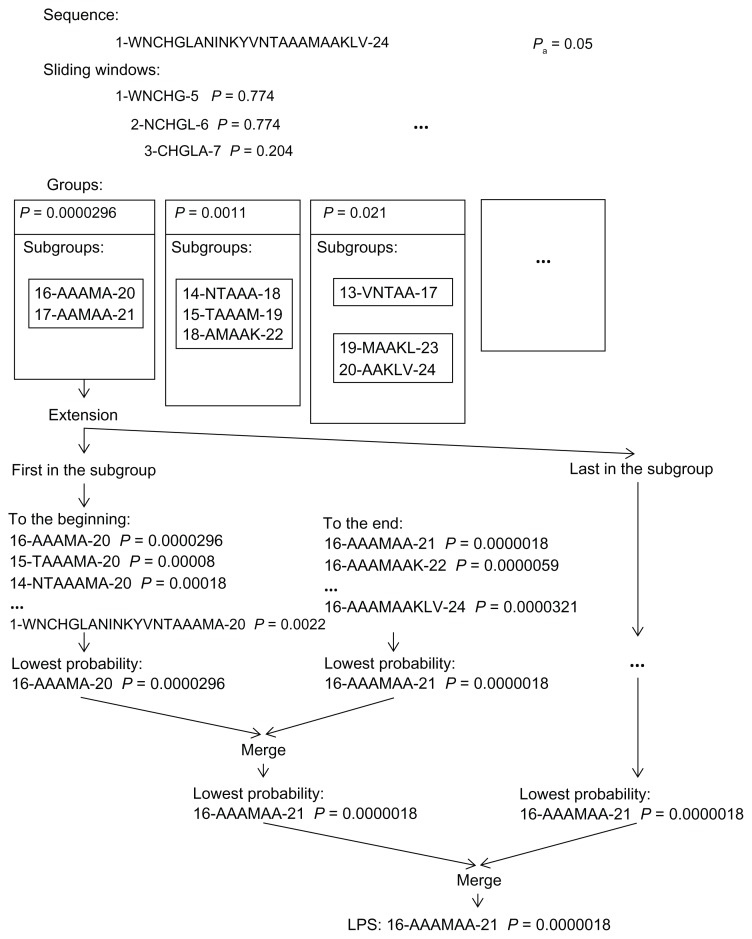
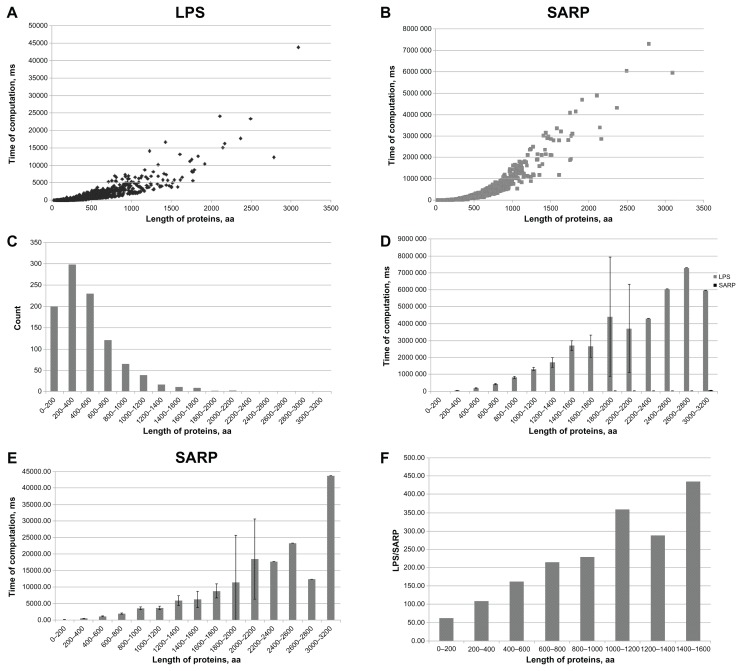
The composition of a defined set of subunits (nucleotides, amino acids) is one of the key features of biological sequences. Compositional biases are local shifts in amino acid or nucleotide frequencies that can occur as an adaptation of an organism to an extreme ecological niche, or as the signature of a specific function or localization of the corresponding protein. The calculation of probability is a method for annotating compositional bias and providing accurate detection of biased subsequences. Here, we present a Sequence Analysis based on the Ranking of Probabilities (SARP), a novel algorithm for the annotation of compositional biases based on ranking subsequences by their probabilities. SARP provides the same accuracy as the previously published Lower Probability Subsequences (LPS) algorithm but performs at an approximately 230-fold faster rate. It can be recommended for use when working with large datasets to reduce the time and resources required.
定义的一组亚基(核苷酸、氨基酸)的组成是生物序列的关键特征之一。组成偏差是氨基酸或核苷酸频率的局部偏移,可以作为生物体对极端生态位的适应,或者作为特定功能或相应蛋白质定位的特征。概率计算是一种注释组成偏差并提供偏置子序列的准确检测的方法。在这里,我们提出了一种基于概率排序的序列分析(SARP),这是一种基于概率对组成偏差进行注释的新算法。SARP 提供了与先前发布的低概率子序列(LPS)算法相同的准确性,但速度快约 230 倍。当处理大型数据集以减少所需的时间和资源时,推荐使用它。