Institute for Systems Biology, Seattle, Washington, United States of America.
PLoS Comput Biol. 2013;9(7):e1003148. doi: 10.1371/journal.pcbi.1003148. Epub 2013 Jul 25.
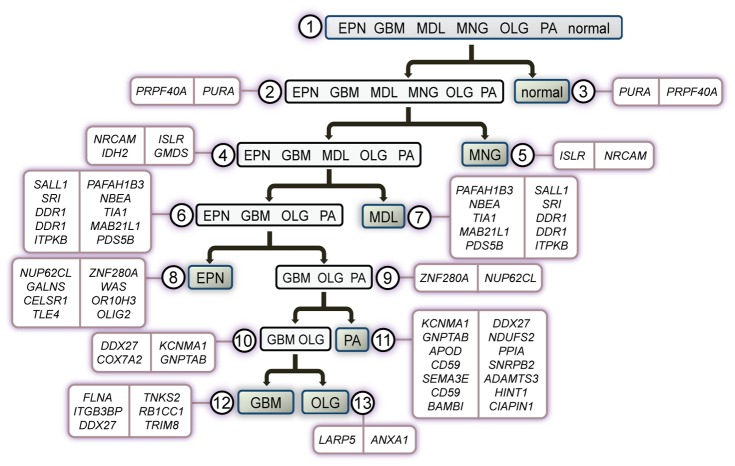
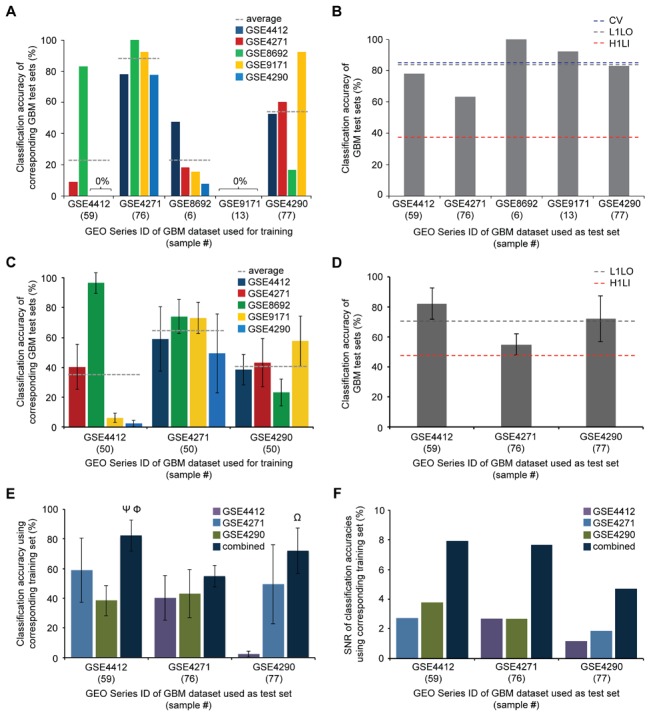
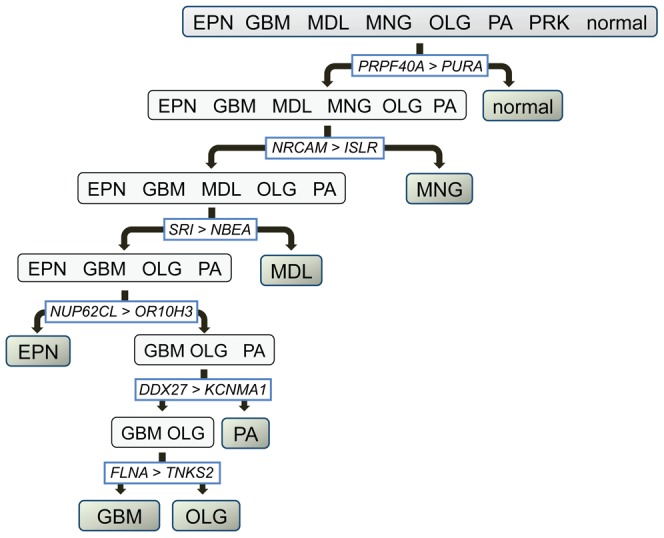
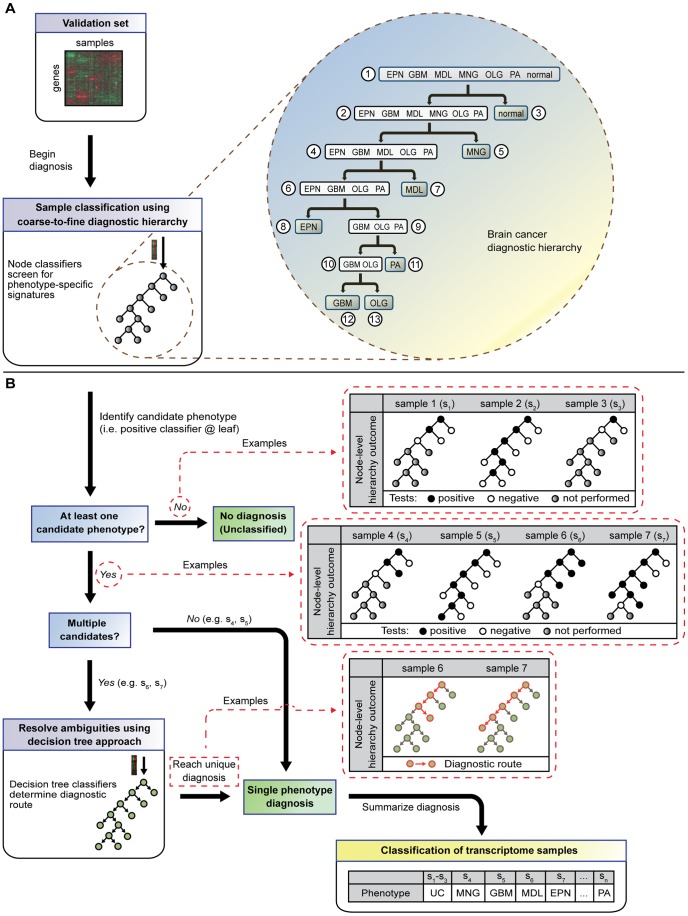
We utilized abundant transcriptomic data for the primary classes of brain cancers to study the feasibility of separating all of these diseases simultaneously based on molecular data alone. These signatures were based on a new method reported herein--Identification of Structured Signatures and Classifiers (ISSAC)--that resulted in a brain cancer marker panel of 44 unique genes. Many of these genes have established relevance to the brain cancers examined herein, with others having known roles in cancer biology. Analyses on large-scale data from multiple sources must deal with significant challenges associated with heterogeneity between different published studies, for it was observed that the variation among individual studies often had a larger effect on the transcriptome than did phenotype differences, as is typical. For this reason, we restricted ourselves to studying only cases where we had at least two independent studies performed for each phenotype, and also reprocessed all the raw data from the studies using a unified pre-processing pipeline. We found that learning signatures across multiple datasets greatly enhanced reproducibility and accuracy in predictive performance on truly independent validation sets, even when keeping the size of the training set the same. This was most likely due to the meta-signature encompassing more of the heterogeneity across different sources and conditions, while amplifying signal from the repeated global characteristics of the phenotype. When molecular signatures of brain cancers were constructed from all currently available microarray data, 90% phenotype prediction accuracy, or the accuracy of identifying a particular brain cancer from the background of all phenotypes, was found. Looking forward, we discuss our approach in the context of the eventual development of organ-specific molecular signatures from peripheral fluids such as the blood.
我们利用丰富的脑癌主要类型转录组数据,研究了仅基于分子数据同时分离所有这些疾病的可行性。这些特征基于本文报道的一种新方法——结构特征和分类器识别(ISSAC),该方法产生了一个包含 44 个独特基因的脑癌标志物面板。这些基因中的许多与本文中检查的脑癌具有相关性,而其他基因在癌症生物学中具有已知作用。对来自多个来源的大规模数据的分析必须应对与不同已发表研究之间异质性相关的重大挑战,因为观察到个体研究之间的变异通常对转录组的影响比对表型差异的影响更大,这是典型的。出于这个原因,我们仅限于研究每个表型至少有两个独立研究的情况,并且还使用统一的预处理管道重新处理了所有研究的原始数据。我们发现,跨多个数据集学习特征极大地提高了在真正独立验证集上的预测性能的可重复性和准确性,即使保持训练集的大小相同。这很可能是由于元特征包含了更多来自不同来源和条件的异质性,同时放大了表型重复的全局特征的信号。当从所有可用的微阵列数据构建脑癌的分子特征时,发现 90%的表型预测准确性,或从所有表型背景中识别特定脑癌的准确性。展望未来,我们将在从血液等外周体液中开发器官特异性分子特征的最终背景下讨论我们的方法。