Laboratory of Virus Evolution, Program in Emerging Infectious Diseases, Duke-NUS Graduate Medical School, Singapore.
PLoS Pathog. 2013;9(8):e1003570. doi: 10.1371/journal.ppat.1003570. Epub 2013 Aug 29.
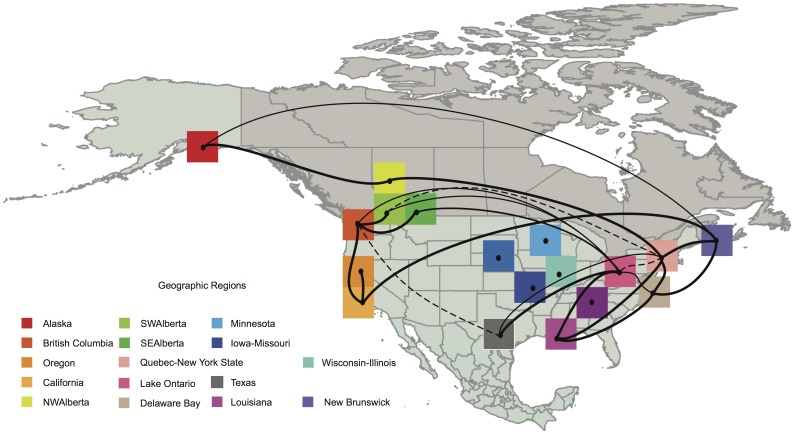
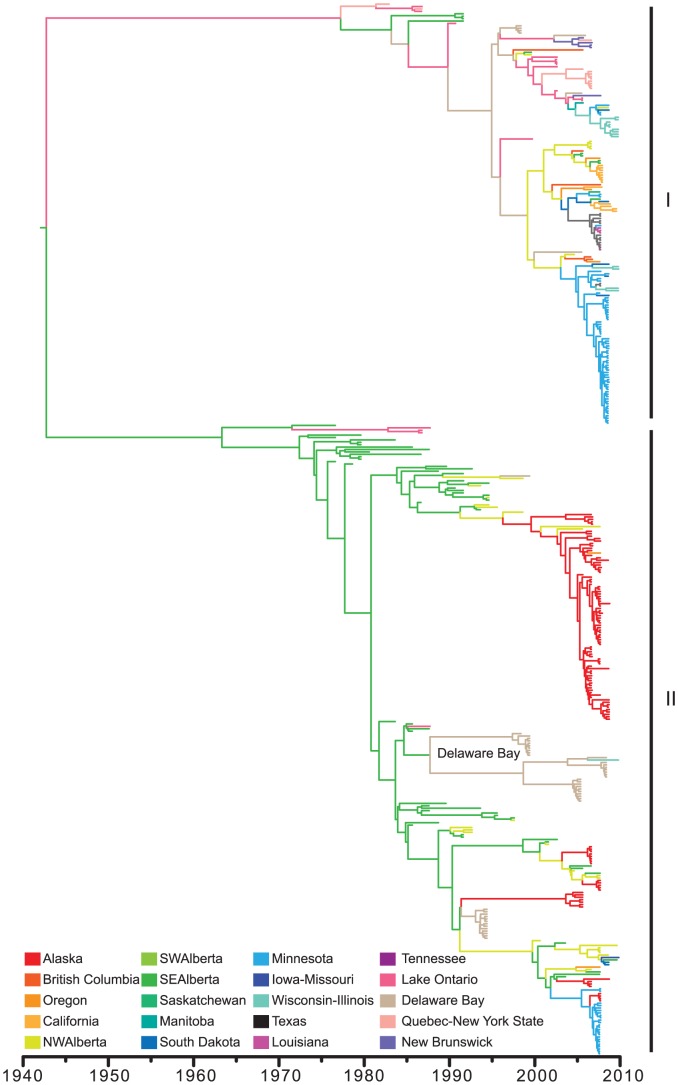
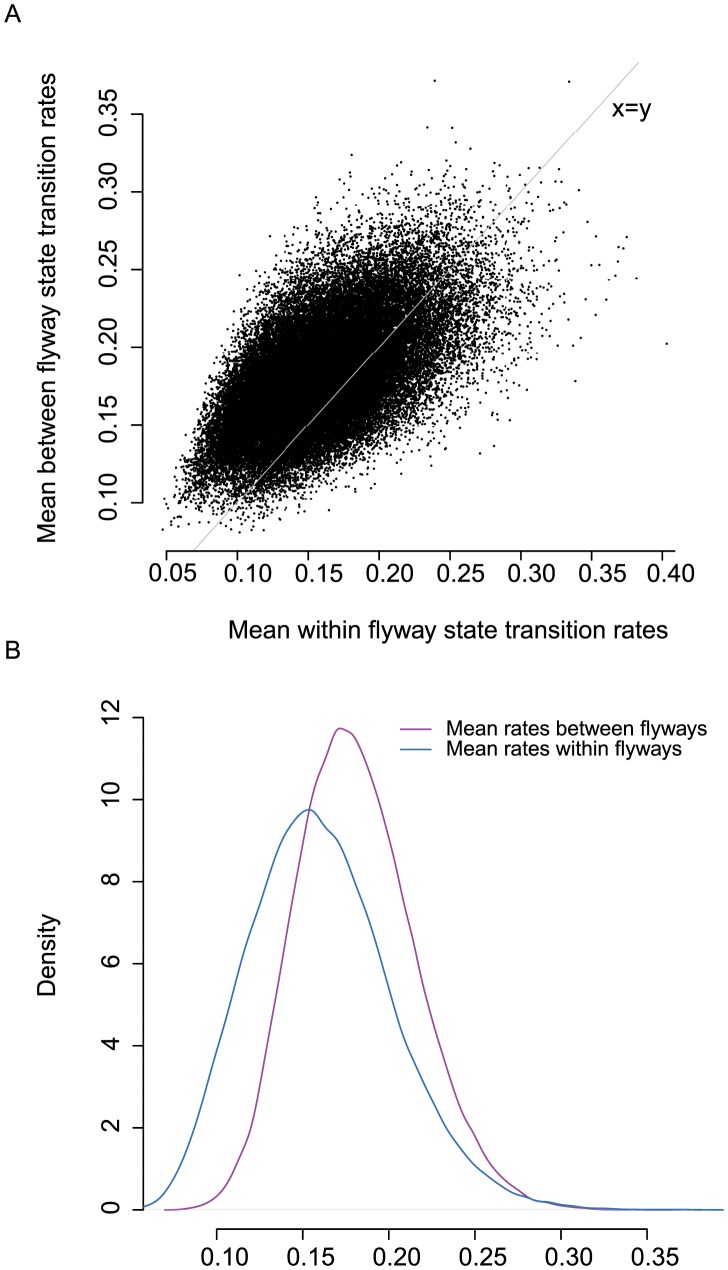
Wild birds have been implicated in the emergence of human and livestock influenza. The successful prediction of viral spread and disease emergence, as well as formulation of preparedness plans have been hampered by a critical lack of knowledge of viral movements between different host populations. The patterns of viral spread and subsequent risk posed by wild bird viruses therefore remain unpredictable. Here we analyze genomic data, including 287 newly sequenced avian influenza A virus (AIV) samples isolated over a 34-year period of continuous systematic surveillance of North American migratory birds. We use a Bayesian statistical framework to test hypotheses of viral migration, population structure and patterns of genetic reassortment. Our results reveal that despite the high prevalence of Charadriiformes infected in Delaware Bay this host population does not appear to significantly contribute to the North American AIV diversity sampled in Anseriformes. In contrast, influenza viruses sampled from Anseriformes in Alberta are representative of the AIV diversity circulating in North American Anseriformes. While AIV may be restricted to specific migratory flyways over short time frames, our large-scale analysis showed that the long-term persistence of AIV was independent of bird flyways with migration between populations throughout North America. Analysis of long-term surveillance data provides vital insights to develop appropriately informed predictive models critical for pandemic preparedness and livestock protection.
野生鸟类被认为是人类和家畜流感的起源。由于对不同宿主种群之间病毒传播的了解甚少,成功预测病毒传播和疾病爆发以及制定防范计划受到了严重阻碍。因此,野生鸟类病毒的传播模式及其随后带来的风险仍然难以预测。在这里,我们分析了基因组数据,包括在对北美候鸟进行连续系统监测的 34 年期间分离的 287 个新测序的甲型流感病毒 (AIV) 样本。我们使用贝叶斯统计框架来检验病毒迁移、种群结构和遗传重配模式的假设。我们的研究结果表明,尽管在特拉华湾感染的Charadriiformes数量很高,但该宿主种群似乎并没有显著促进在anseriformes 中采样的北美 AIV 多样性。相比之下,从艾伯塔省anseriformes 中采样的流感病毒代表了在北美anseriformes 中循环的 AIV 多样性。虽然 AIV 可能在短时间内受到特定迁徙飞行路线的限制,但我们的大规模分析表明,AIV 的长期存在与鸟类迁徙无关,而是与整个北美地区的种群间迁徙有关。对长期监测数据的分析为制定适当知情的预测模型提供了重要的见解,这些模型对于大流行防范和牲畜保护至关重要。