Islam Md Fahmid, Hoque Md Moinul, Banik Rajat Suvra, Roy Sanjoy, Sumi Sharmin Sultana, Hassan F M Nazmul, Tomal Md Tauhid Siddiki, Ullah Ahmad, Rahman K M Taufiqur
Biotechnology and Genetic Engineering Discipline, Khulna University, Khulna 9208, Bangladesh.
Forestry and Wood Technology Discipline, Khulna University, Khulna 9208, Bangladesh.
J Clin Bioinforma. 2013 Oct 6;3(1):19. doi: 10.1186/2043-9113-3-19.
Large scale understanding of complex and dynamic alterations in cellular and subcellular levels during cancer in contrast to normal condition has facilitated the emergence of sophisticated systemic approaches like network biology in recent times. As most biological networks show modular properties, the analysis of differential modularity between normal and cancer protein interaction networks can be a good way to understand cancer more significantly. Two aspects of biological network modularity e.g. detection of molecular complexes (potential modules or clusters) and identification of crucial nodes forming the overlapping modules have been considered in this regard.
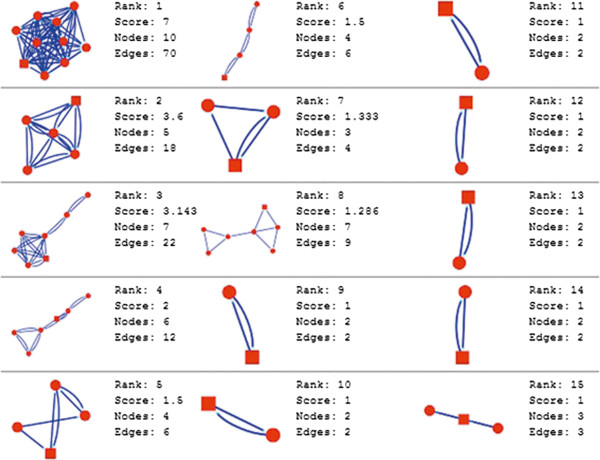
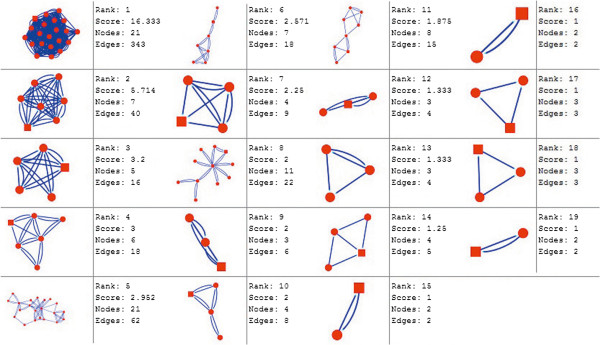
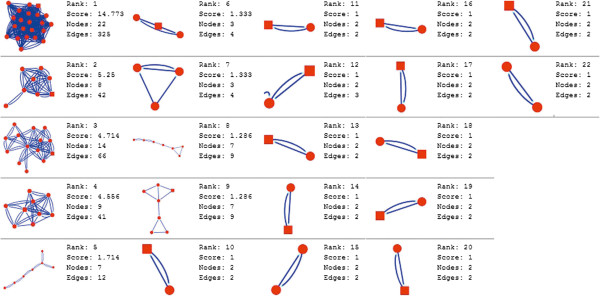
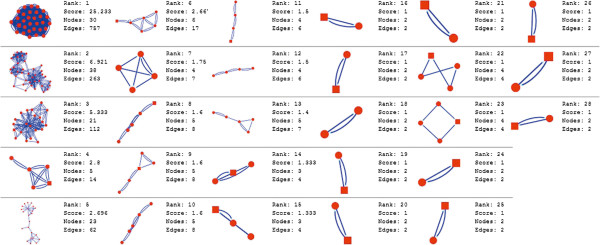
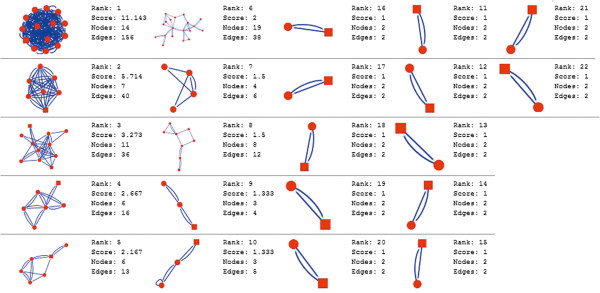
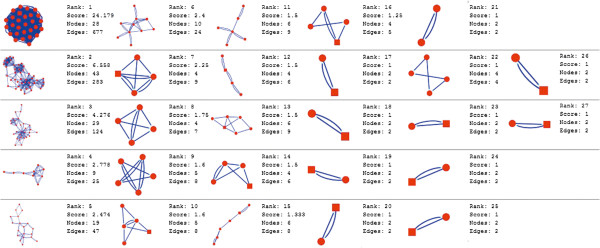
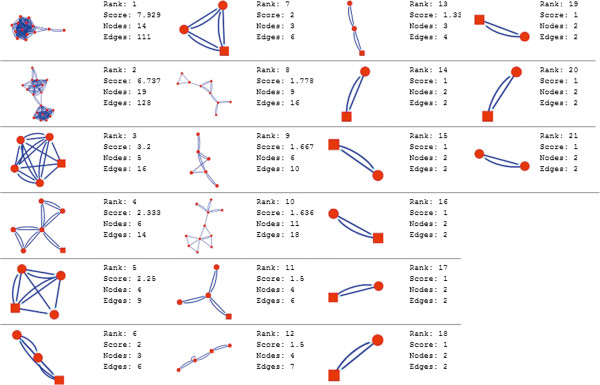
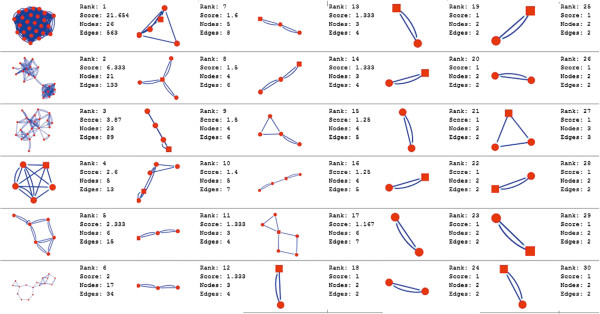
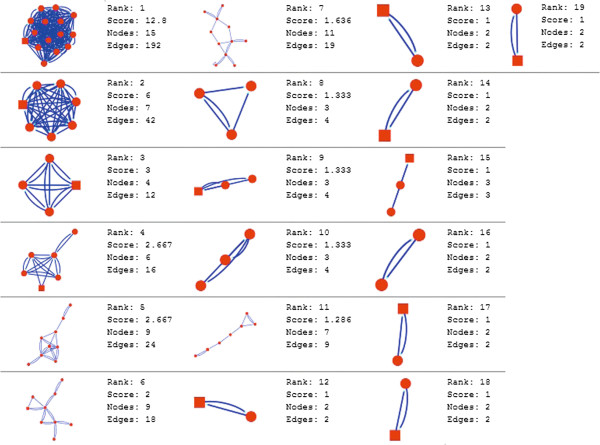
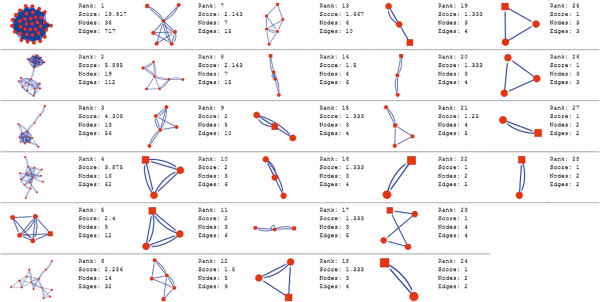
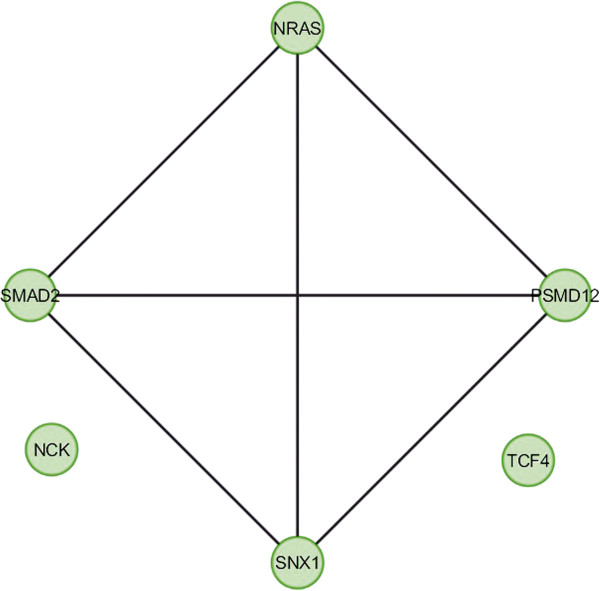
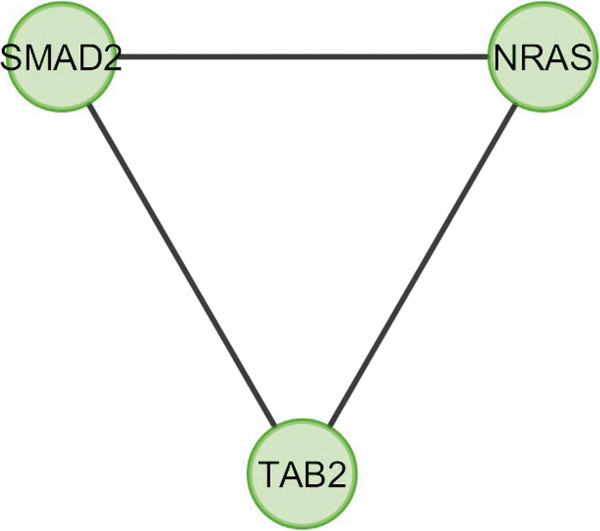
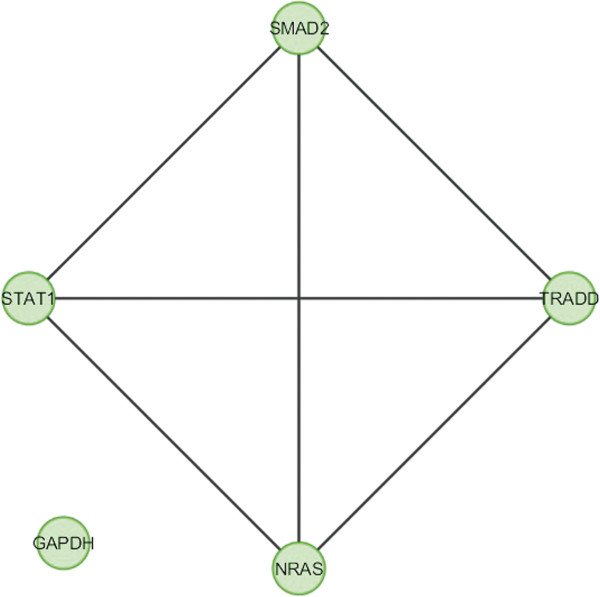
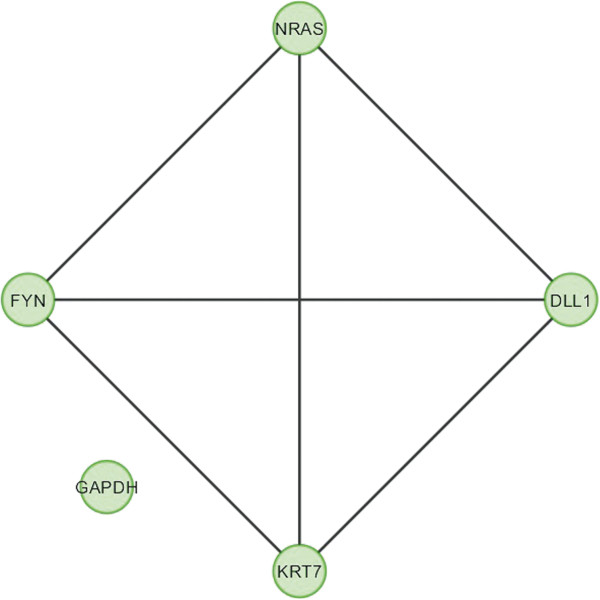
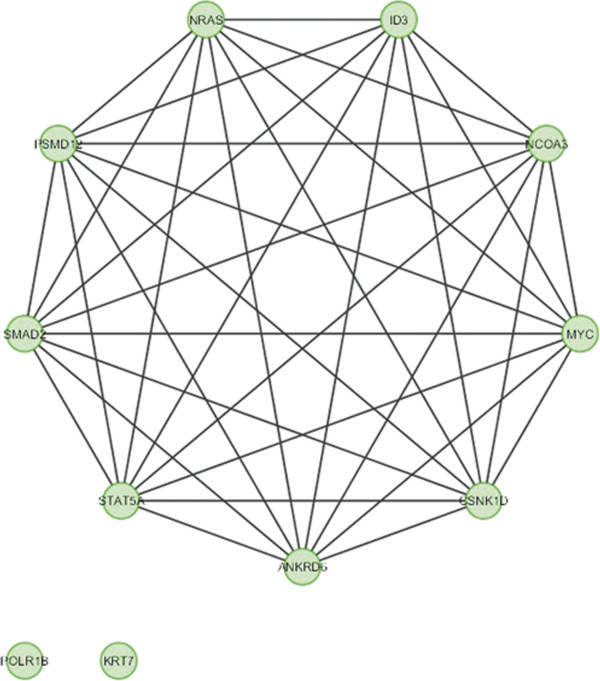
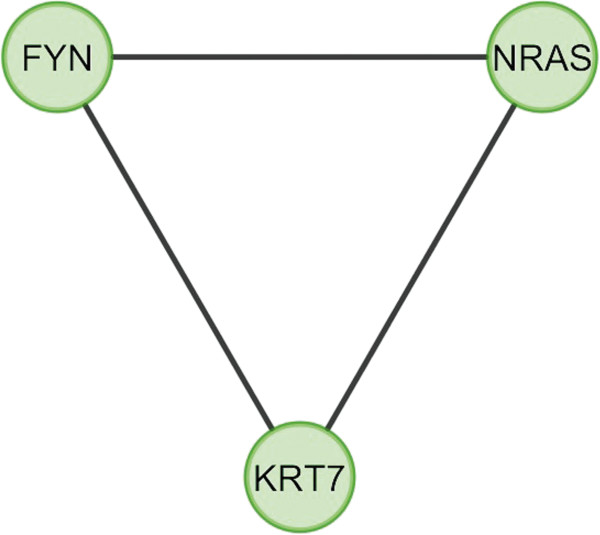
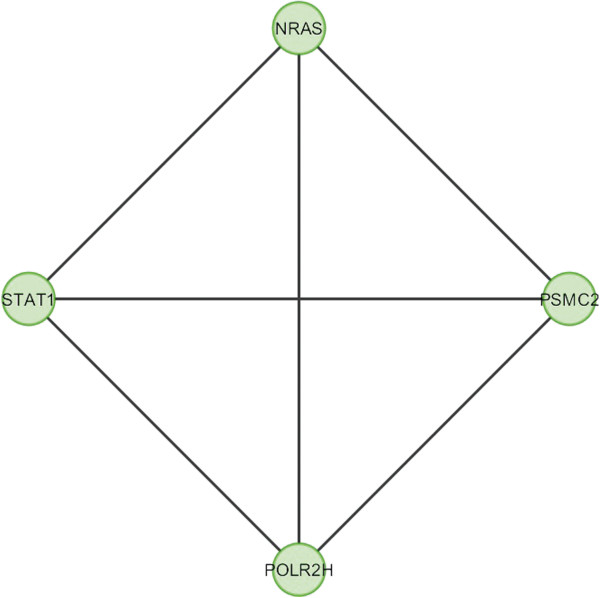
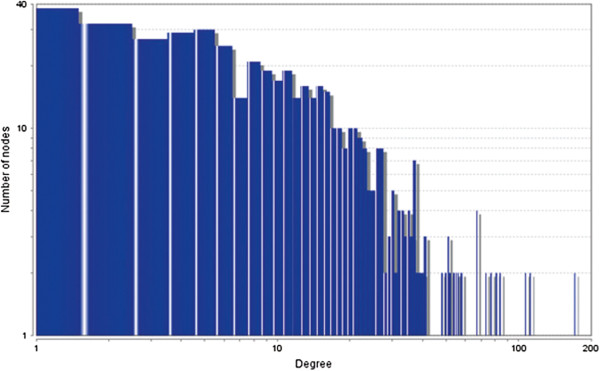
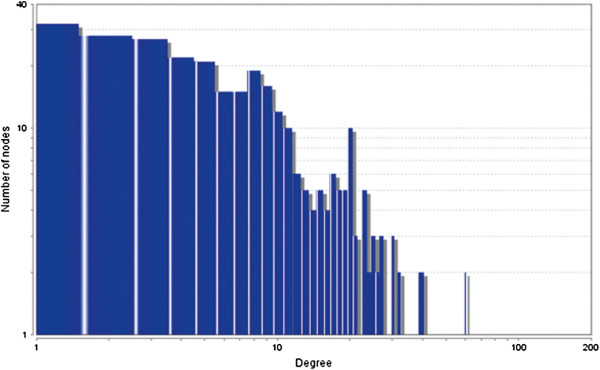
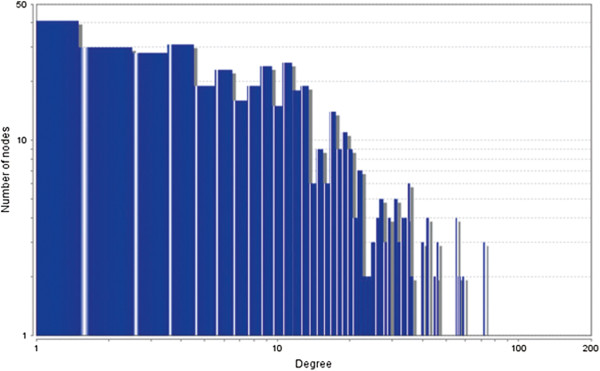
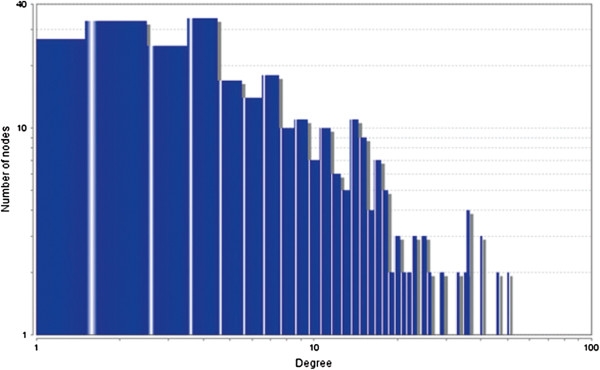
In the current study, the computational analysis of previously published protein interaction networks (PINs) has been conducted to identify the molecular complexes and crucial nodes of the networks. Protein molecules involved in ten major cancer signal transduction pathways were used to construct the networks based on expression data of five tissues e.g. bone, breast, colon, kidney and liver in both normal and cancer conditions. MCODE (molecular complex detection) and ModuLand methods have been used to identify the molecular complexes and crucial nodes of the networks respectively.
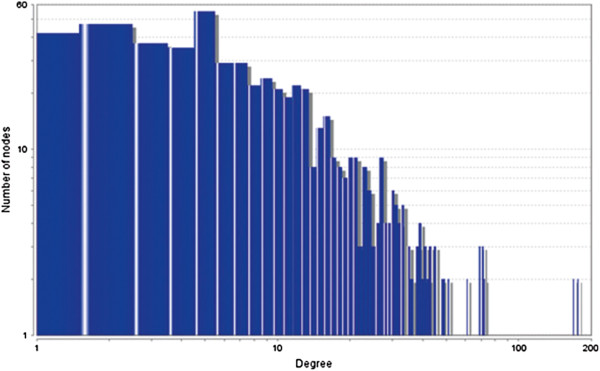
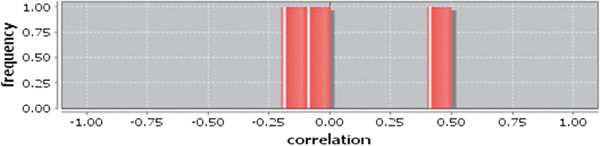
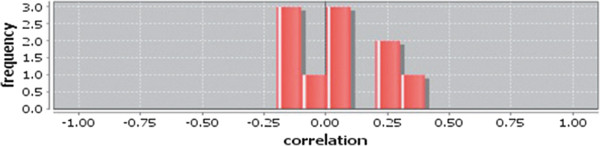
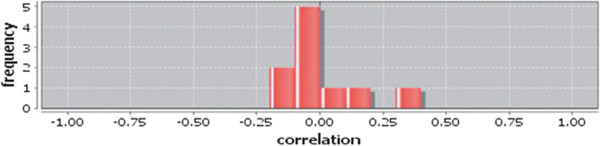
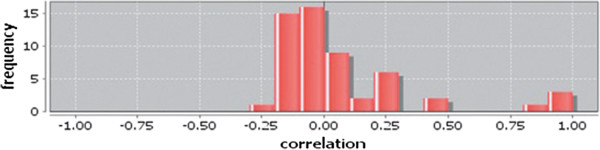
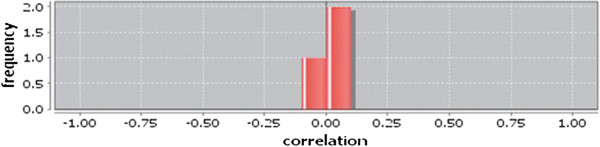
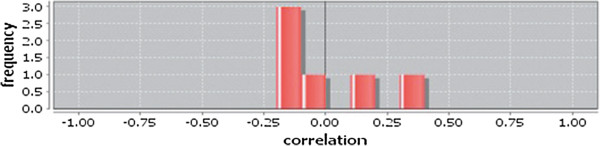
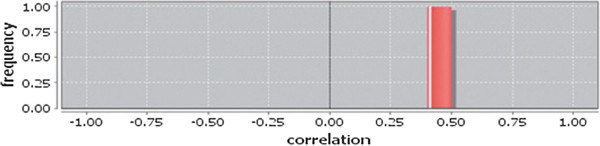
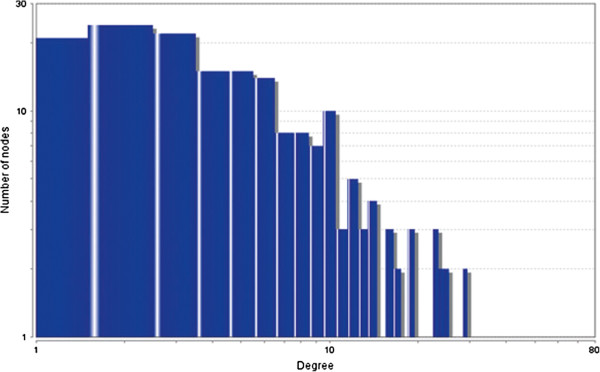
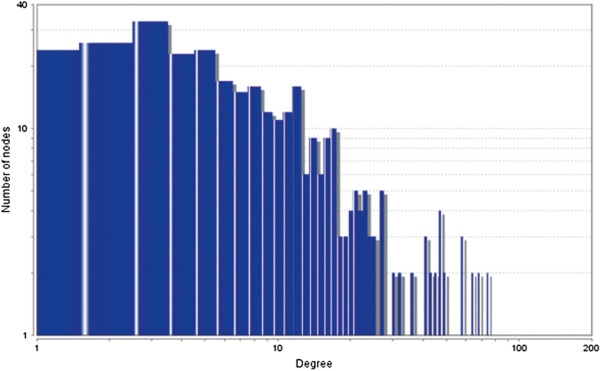
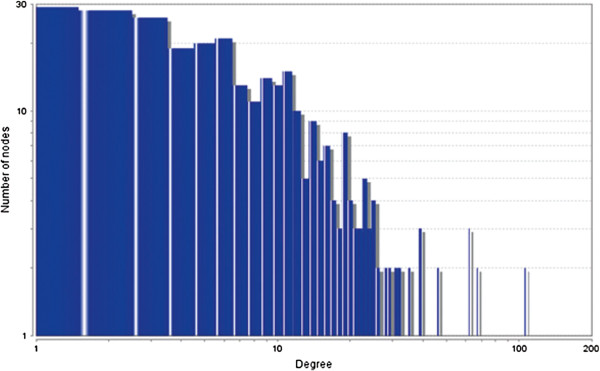
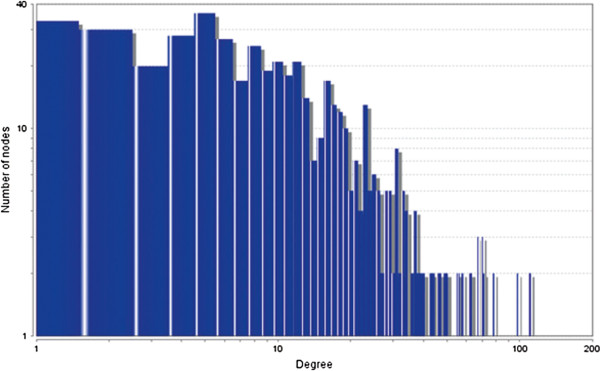
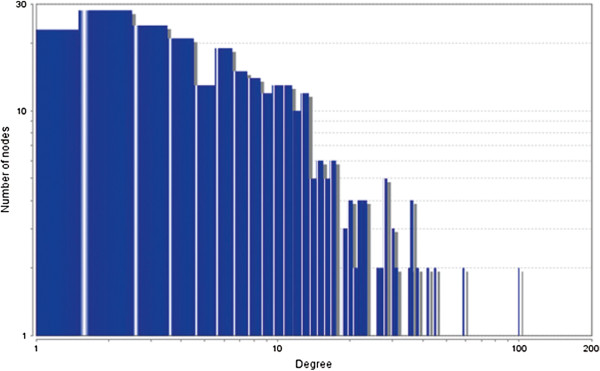
In case of all tissues, cancer PINs show higher level of clustering (formation of molecular complexes) than the normal ones. In contrast, lower level modular overlapping is found in cancer PINs than the normal ones. Thus a proposition can be made regarding the formation of some giant nodes in the cancer networks with very high degree and resulting in reduced overlapping among the network modules though the predicted molecular complex numbers are higher in cancer conditions.
The study predicts some major molecular complexes that might act as the important regulators in cancer progression. The crucial nodes identified in this study can be potential drug targets to combat cancer.
与正常情况相比,大规模了解癌症发生过程中细胞和亚细胞水平上复杂且动态的变化,推动了诸如网络生物学等精密系统方法在近期的出现。由于大多数生物网络具有模块化特性,分析正常与癌症蛋白质相互作用网络之间的差异模块化,可能是更深入理解癌症的一个好方法。在这方面,已经考虑了生物网络模块化的两个方面,即分子复合物(潜在模块或簇)的检测以及形成重叠模块的关键节点的识别。
在本研究中,对先前发表的蛋白质相互作用网络(PINs)进行了计算分析,以识别网络中的分子复合物和关键节点。基于正常和癌症状态下骨、乳腺、结肠、肾脏和肝脏这五种组织的表达数据,使用参与十条主要癌症信号转导途径的蛋白质分子构建网络。分别使用MCODE(分子复合物检测)和ModuLand方法来识别网络中的分子复合物和关键节点。
在所有组织中,癌症PINs的聚类水平(分子复合物的形成)均高于正常PINs。相比之下,癌症PINs中的模块化重叠水平低于正常PINs。因此,可以提出这样一个观点,即癌症网络中形成了一些度非常高的巨型节点,尽管预测的癌症状态下分子复合物数量较多,但却导致网络模块之间的重叠减少。
该研究预测了一些可能在癌症进展中起重要调节作用的主要分子复合物。本研究中识别出的关键节点可能是抗癌的潜在药物靶点。