Department of Epidemiology, Harvard School of Public Health, Boston, Massachusetts, United States of America ; Center for Communicable Disease Dynamics, Harvard School of Public Health, Boston, Massachusetts, United States of America.
PLoS Comput Biol. 2013;9(10):e1003268. doi: 10.1371/journal.pcbi.1003268. Epub 2013 Oct 10.
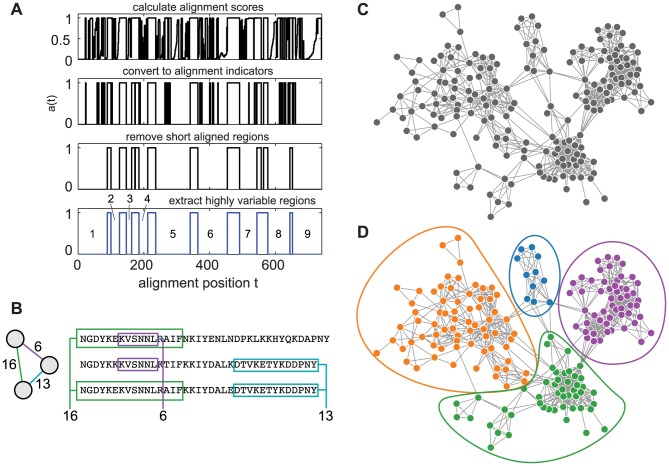
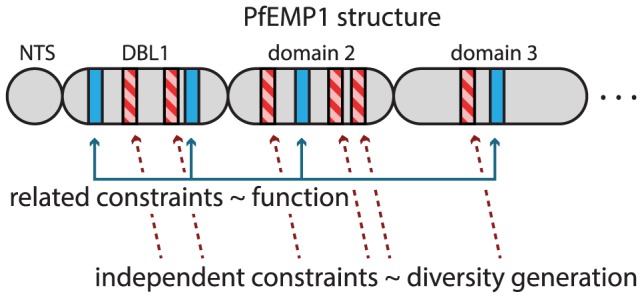
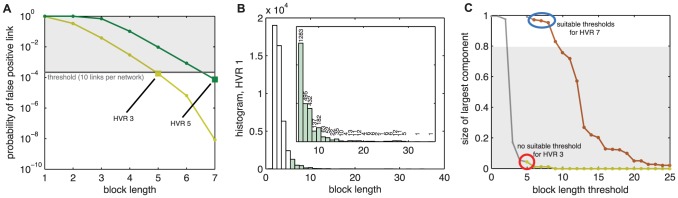
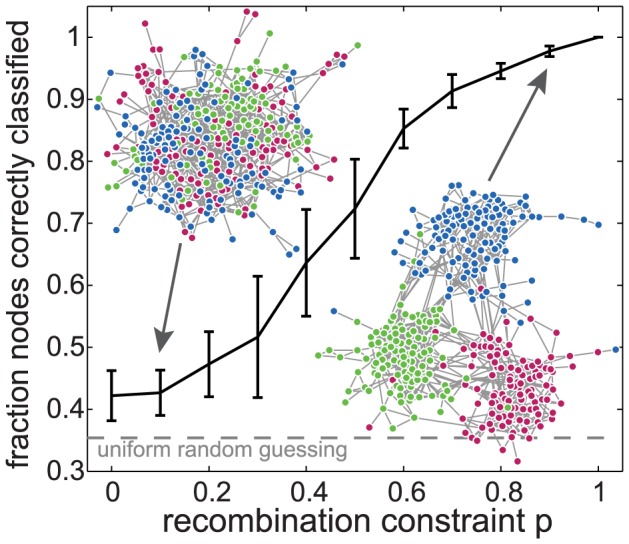
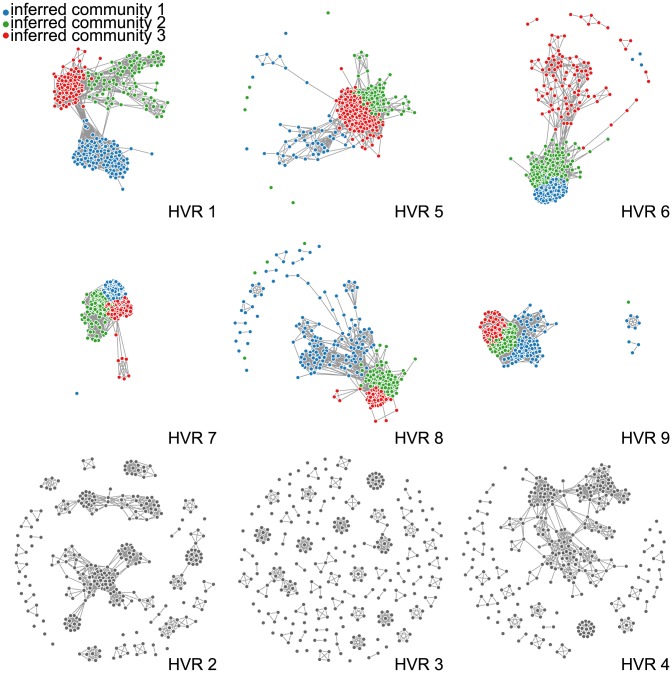
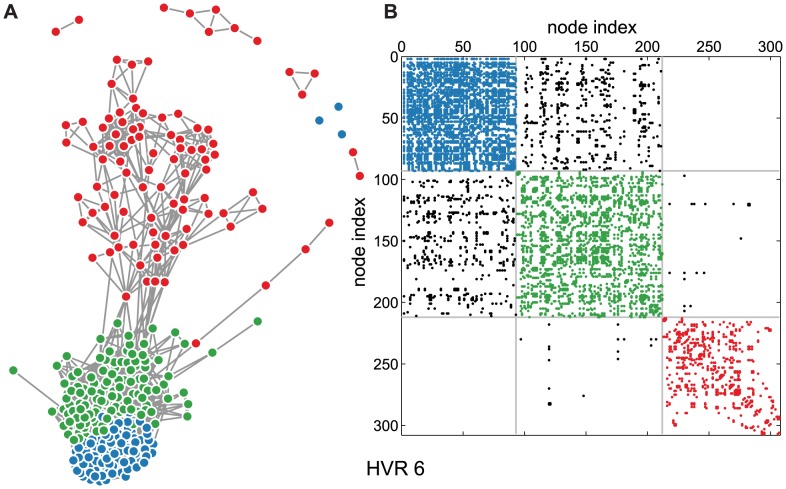
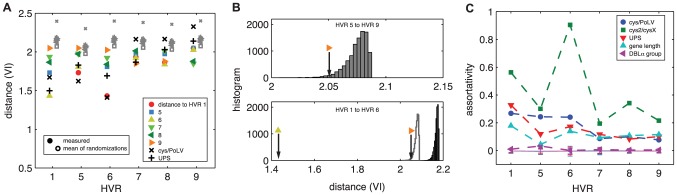
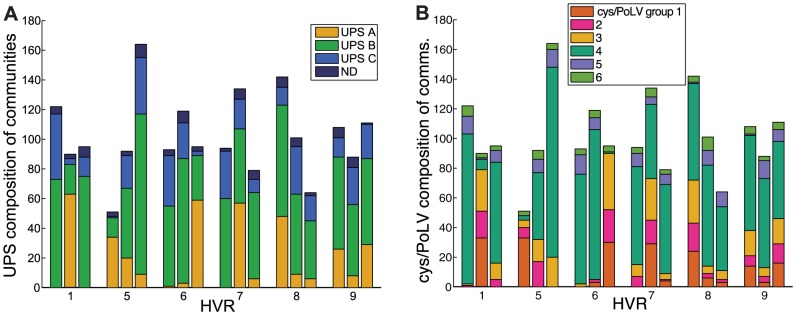
The var genes of the human malaria parasite Plasmodium falciparum present a challenge to population geneticists due to their extreme diversity, which is generated by high rates of recombination. These genes encode a primary antigen protein called PfEMP1, which is expressed on the surface of infected red blood cells and elicits protective immune responses. Var gene sequences are characterized by pronounced mosaicism, precluding the use of traditional phylogenetic tools that require bifurcating tree-like evolutionary relationships. We present a new method that identifies highly variable regions (HVRs), and then maps each HVR to a complex network in which each sequence is a node and two nodes are linked if they share an exact match of significant length. Here, networks of var genes that recombine freely are expected to have a uniformly random structure, but constraints on recombination will produce network communities that we identify using a stochastic block model. We validate this method on synthetic data, showing that it correctly recovers populations of constrained recombination, before applying it to the Duffy Binding Like-α (DBLα) domain of var genes. We find nine HVRs whose network communities map in distinctive ways to known DBLα classifications and clinical phenotypes. We show that the recombinational constraints of some HVRs are correlated, while others are independent. These findings suggest that this micromodular structuring facilitates independent evolutionary trajectories of neighboring mosaic regions, allowing the parasite to retain protein function while generating enormous sequence diversity. Our approach therefore offers a rigorous method for analyzing evolutionary constraints in var genes, and is also flexible enough to be easily applied more generally to any highly recombinant sequences.
人类疟疾寄生虫疟原虫的 var 基因由于其高度的多样性,给群体遗传学家带来了挑战,这种多样性是由高重组率产生的。这些基因编码一种主要的抗原蛋白,称为 PfEMP1,它在感染的红细胞表面表达,并引发保护性免疫反应。Var 基因序列的特征是明显的镶嵌性,排除了使用需要二叉树状进化关系的传统系统发育工具。我们提出了一种新的方法,可以识别高度可变区(HVR),然后将每个 HVR 映射到一个复杂的网络中,其中每个序列都是一个节点,如果它们具有显著长度的精确匹配,则两个节点相连。在这里,自由重组的 var 基因网络预计将具有均匀随机的结构,但重组的约束将产生网络社区,我们使用随机块模型来识别这些社区。我们在合成数据上验证了这种方法,表明它可以正确恢复受约束重组的种群,然后将其应用于 var 基因的 Duffy 结合样-α(DBLα)结构域。我们发现了九个 HVR,它们的网络社区以独特的方式映射到已知的 DBLα 分类和临床表型。我们表明,一些 HVR 的重组约束是相关的,而其他则是独立的。这些发现表明,这种微观模块化结构促进了相邻镶嵌区域的独立进化轨迹,使寄生虫在保持蛋白功能的同时产生巨大的序列多样性。因此,我们的方法为分析 var 基因中的进化约束提供了一种严格的方法,并且足够灵活,可以很容易地应用于任何高度重组的序列。