Abergel Chantal
Information Génomique et Structurale, IGS UMR 7256, CNRS, Aix-Marseille Université, IMM, FR3479, 163 Avenue de Luminy - case 934, 13288 Marseille CEDEX 09, France.
Acta Crystallogr D Biol Crystallogr. 2013 Nov;69(Pt 11):2167-73. doi: 10.1107/S0907444913015291. Epub 2013 Oct 12.
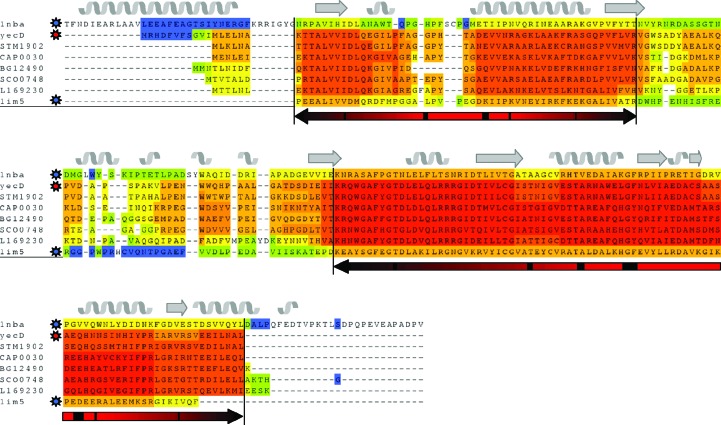
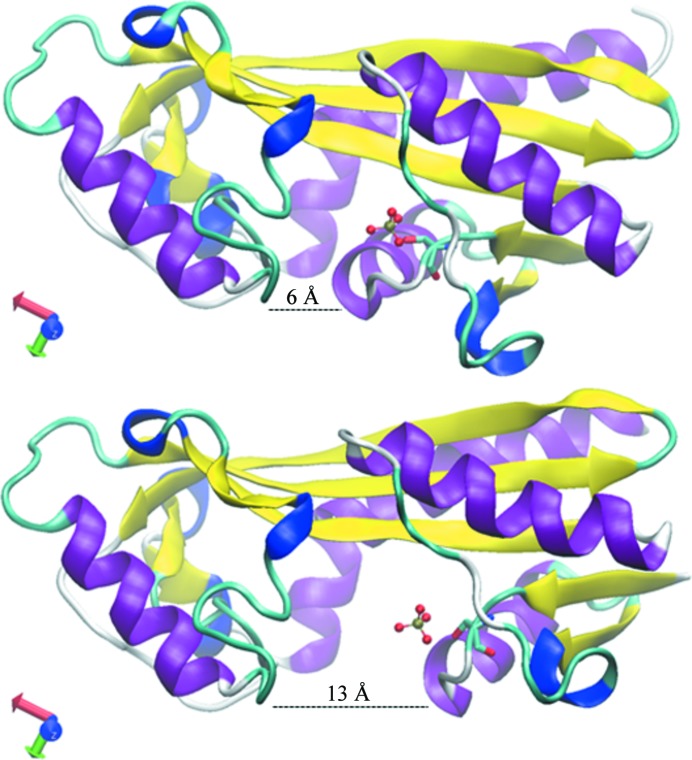
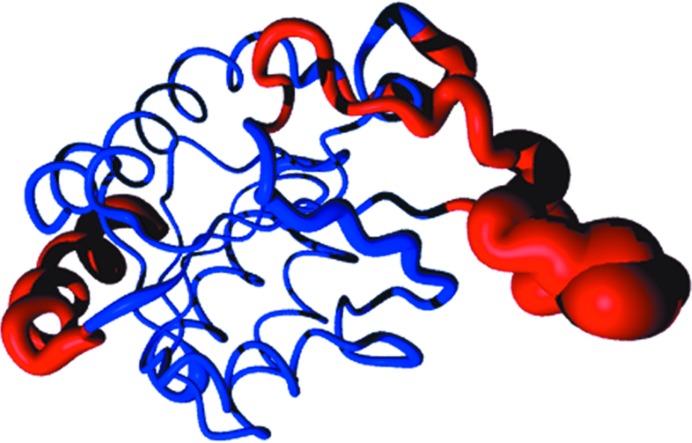
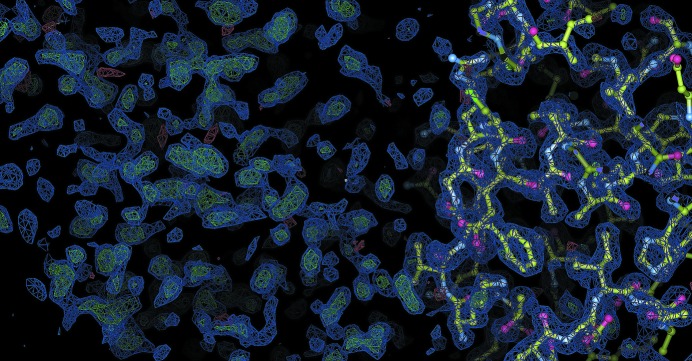
Molecular replacement is the method of choice for X-ray crystallographic structure determination provided that suitable structural homologues are available in the PDB. Presently, there are ~80,000 structures in the PDB (8074 were deposited in the year 2012 alone), of which ~70% have been solved by molecular replacement. For successful molecular replacement the model must cover at least 50% of the total structure and the Cα r.m.s.d. between the core model and the structure to be solved must be less than 2 Å. Here, an approach originally implemented in the CaspR server (http://www.igs.cnrs-mrs.fr/Caspr2/index.cgi) based on homology modelling to search for a molecular-replacement solution is discussed. How the use of as much information as possible from different sources can improve the model(s) is briefly described. The combination of structural information with distantly related sequences is crucial to optimize the multiple alignment that will define the boundaries of the core domains. PDB clusters (sequences with ≥30% identical residues) can also provide information on the eventual changes in conformation and will help to explore the relative orientations assumed by protein subdomains. Normal-mode analysis can also help in generating series of conformational models in the search for a molecular-replacement solution. Of course, finding a correct solution is only the first step and the accuracy of the identified solution is as important as the data quality to proceed through refinement. Here, some possible reasons for failure are discussed and solutions are proposed using a set of successful examples.
如果蛋白质数据银行(PDB)中有合适的结构同源物,分子置换是X射线晶体学结构测定的首选方法。目前,PDB中有约80,000个结构(仅在2012年就存入了8074个),其中约70%是通过分子置换解析出来的。为了成功进行分子置换,模型必须覆盖至少50%的整个结构,并且核心模型与待解析结构之间的Cα均方根偏差(r.m.s.d.)必须小于2 Å。本文讨论了一种最初在CaspR服务器(http://www.igs.cnrs-mrs.fr/Caspr2/index.cgi)中实现的基于同源建模来寻找分子置换解决方案的方法。简要描述了如何利用来自不同来源的尽可能多的信息来改进模型。将结构信息与远缘相关序列相结合对于优化多序列比对至关重要,多序列比对将定义核心结构域的边界。PDB聚类(具有≥30%相同残基的序列)也可以提供有关最终构象变化的信息,并有助于探索蛋白质亚结构域假定的相对取向。正常模式分析也有助于在寻找分子置换解决方案时生成一系列构象模型。当然,找到正确的解决方案只是第一步,并且所确定解决方案的准确性与数据质量对于进行精修同样重要。本文讨论了一些可能导致失败的原因,并使用一组成功的例子提出了解决方案。