Computational Biology Program, Memorial Sloan-Kettering Cancer Center, New York, New York, United States of America ; Tri-Institutional Program for Computational Biology and Medicine, Weill Cornell Medical College, New York, New York, United States of America.
Computational Biology Program, Memorial Sloan-Kettering Cancer Center, New York, New York, United States of America.
PLoS Comput Biol. 2013;9(12):e1003290. doi: 10.1371/journal.pcbi.1003290. Epub 2013 Dec 19.
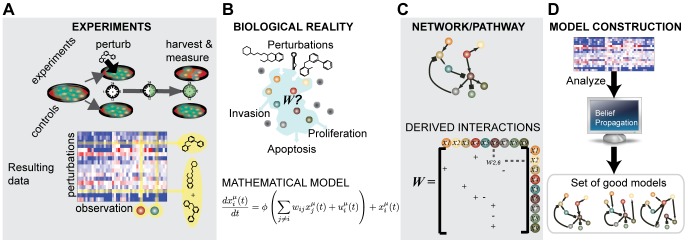
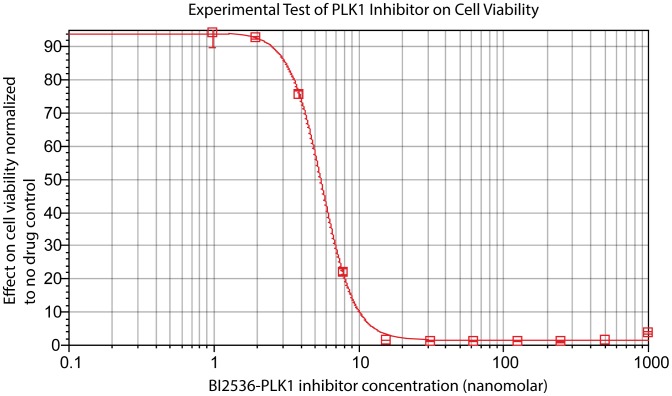
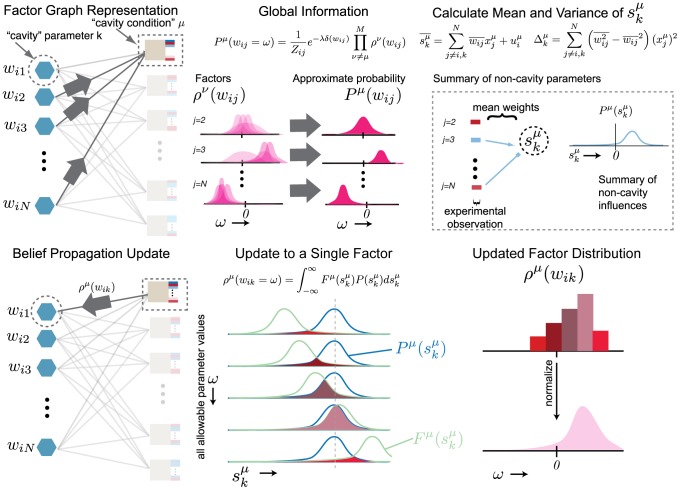
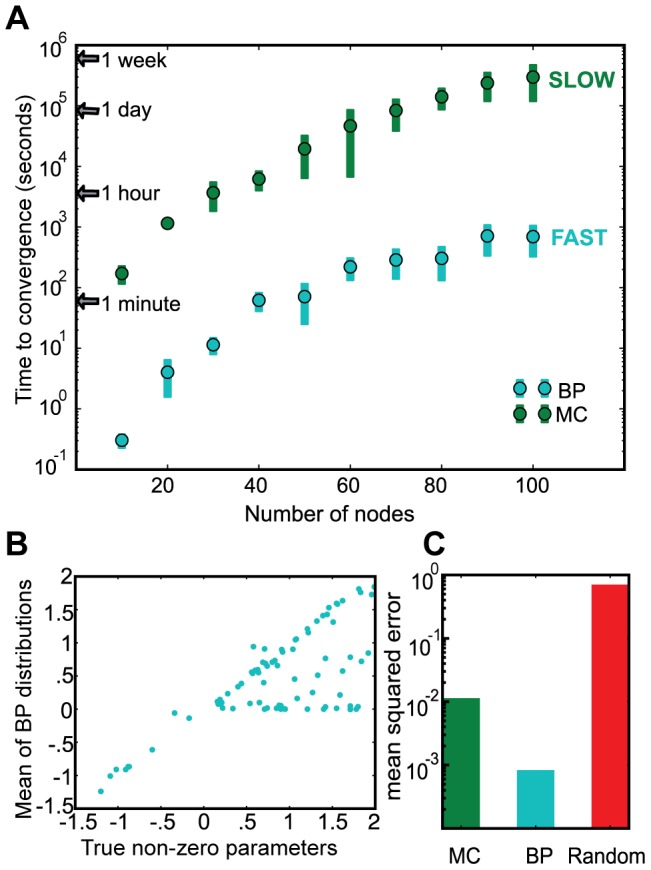
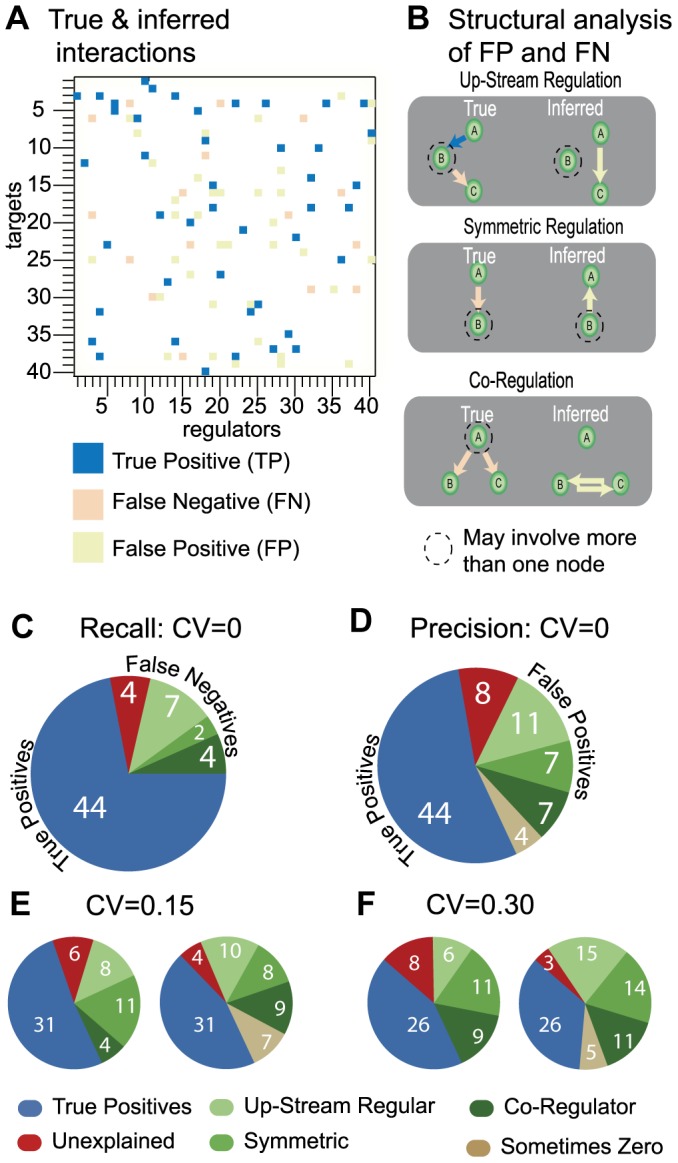
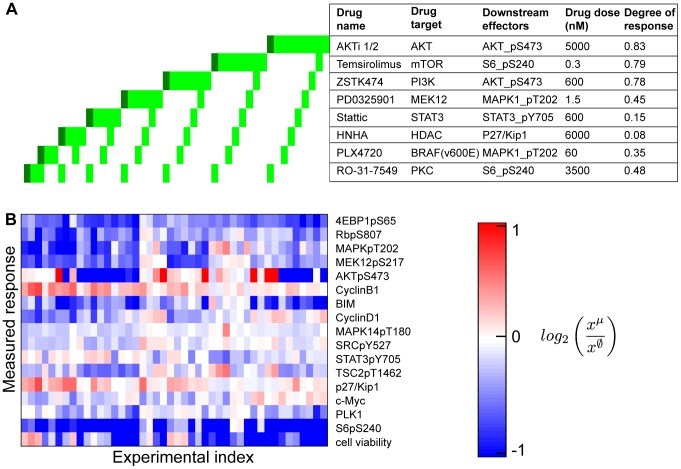
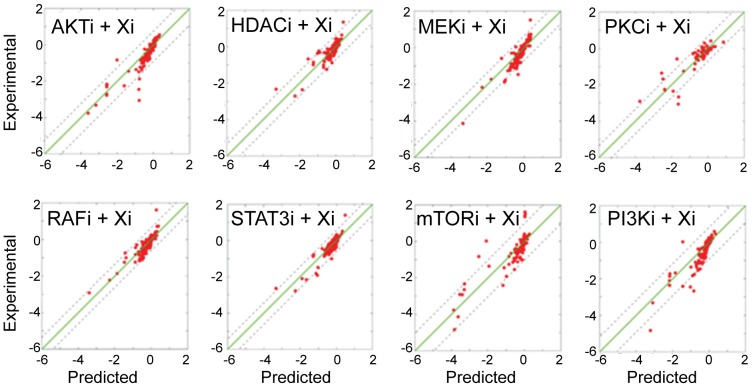
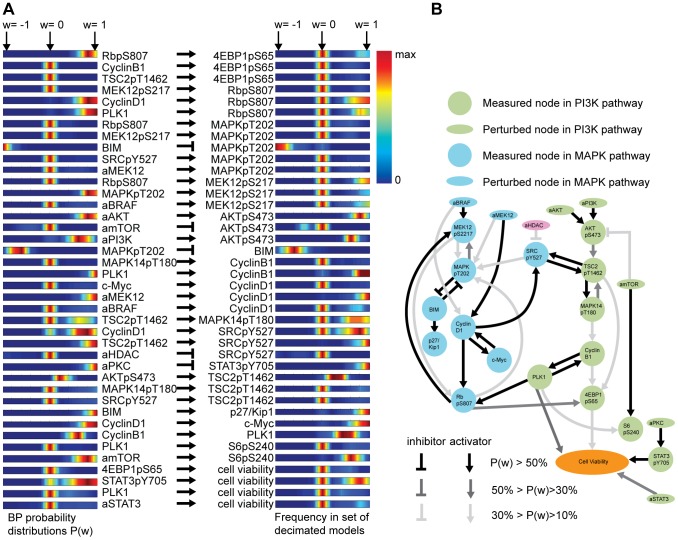
We present a powerful experimental-computational technology for inferring network models that predict the response of cells to perturbations, and that may be useful in the design of combinatorial therapy against cancer. The experiments are systematic series of perturbations of cancer cell lines by targeted drugs, singly or in combination. The response to perturbation is quantified in terms of relative changes in the measured levels of proteins, phospho-proteins and cellular phenotypes such as viability. Computational network models are derived de novo, i.e., without prior knowledge of signaling pathways, and are based on simple non-linear differential equations. The prohibitively large solution space of all possible network models is explored efficiently using a probabilistic algorithm, Belief Propagation (BP), which is three orders of magnitude faster than standard Monte Carlo methods. Explicit executable models are derived for a set of perturbation experiments in SKMEL-133 melanoma cell lines, which are resistant to the therapeutically important inhibitor of RAF kinase. The resulting network models reproduce and extend known pathway biology. They empower potential discoveries of new molecular interactions and predict efficacious novel drug perturbations, such as the inhibition of PLK1, which is verified experimentally. This technology is suitable for application to larger systems in diverse areas of molecular biology.
我们提出了一种强大的实验计算技术,用于推断能够预测细胞对扰动反应的网络模型,这对于设计针对癌症的组合疗法可能是有用的。实验是通过靶向药物对癌细胞系进行的系统的扰动系列,单独或联合使用。扰动的反应是根据所测量的蛋白质、磷酸化蛋白质和细胞表型(如存活率)水平的相对变化来定量的。计算网络模型是从头开始推导的,即没有信号通路的先验知识,并且基于简单的非线性微分方程。使用概率算法信念传播 (BP) 有效地探索了所有可能网络模型的 prohibitively 大的解空间,BP 比标准的蒙特卡罗方法快三个数量级。为 SKMEL-133 黑色素瘤细胞系中的一组扰动实验推导出了明确可执行的模型,这些细胞系对 RAF 激酶抑制剂具有重要的治疗抗性。所得的网络模型再现和扩展了已知的通路生物学。它们使发现新的分子相互作用和预测有效的新型药物扰动成为可能,例如 PLK1 的抑制作用,这在实验中得到了验证。这项技术适用于分子生物学各个领域的更大系统的应用。