Zheng Heping, Hou Jing, Zimmerman Matthew D, Wlodawer Alexander, Minor Wladek
University of Virginia, Department of Molecular Physiology and Biological Physics , 1340 Jefferson Park Avenue, Charlottesville, VA 22908 , USA.
Expert Opin Drug Discov. 2014 Feb;9(2):125-37. doi: 10.1517/17460441.2014.872623. Epub 2013 Dec 28.
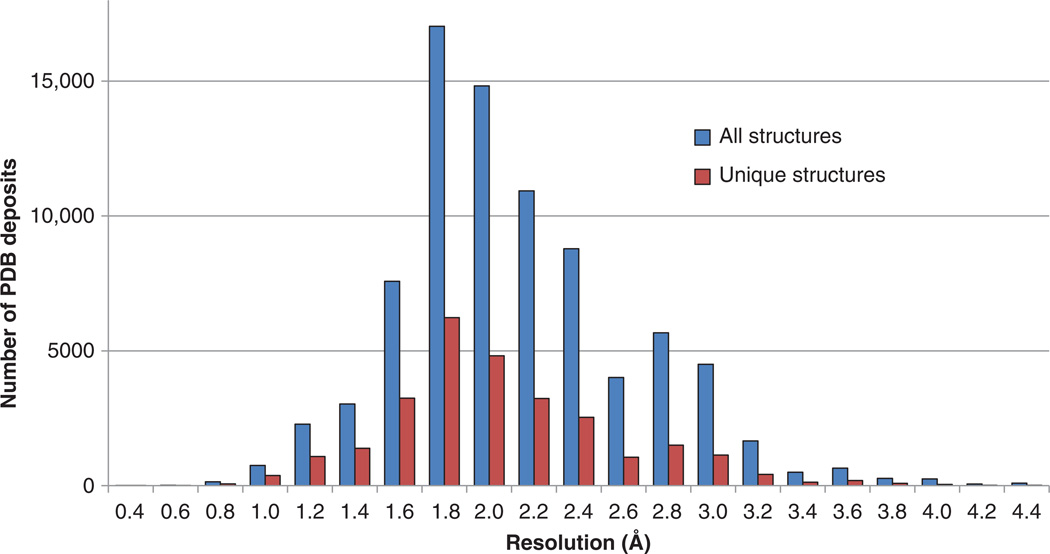
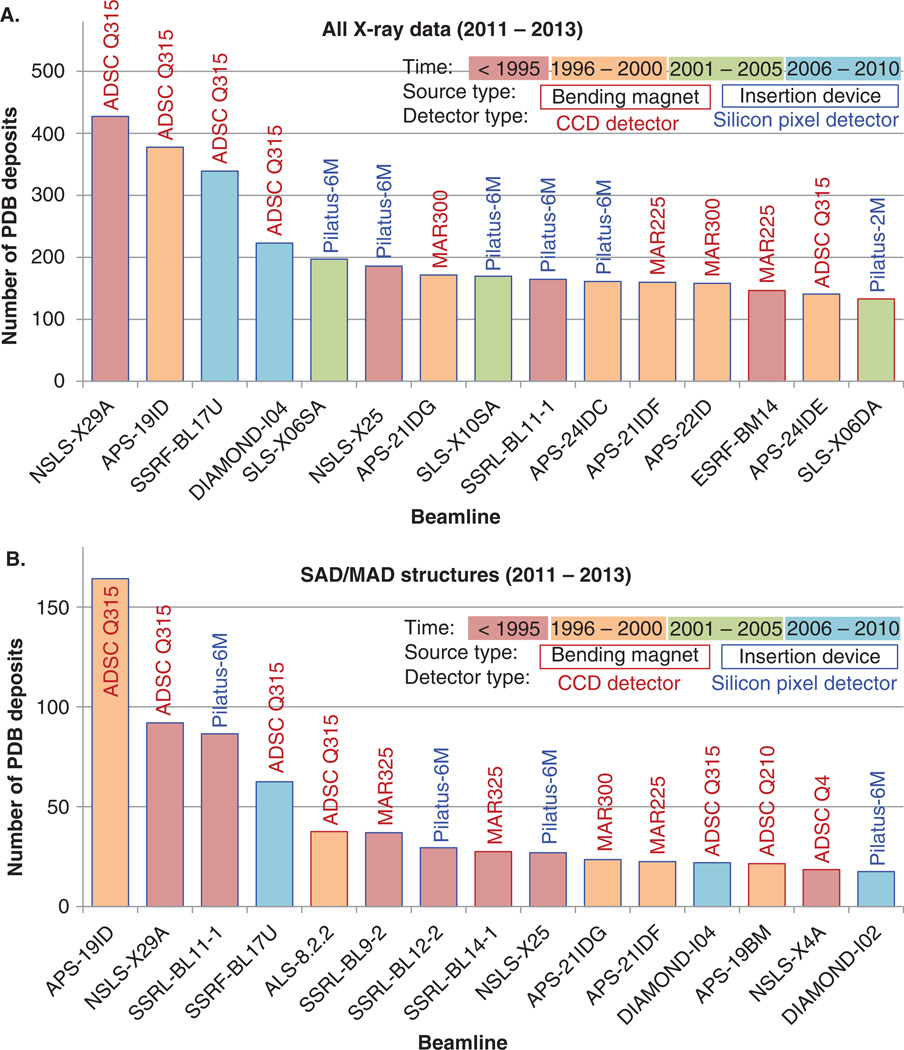
X-ray crystallography plays an important role in structure-based drug design (SBDD), and accurate analysis of crystal structures of target macromolecules and macromolecule-ligand complexes is critical at all stages. However, whereas there has been significant progress in improving methods of structural biology, particularly in X-ray crystallography, corresponding progress in the development of computational methods (such as in silico high-throughput screening) is still on the horizon. Crystal structures can be overinterpreted and thus bias hypotheses and follow-up experiments. As in any experimental science, the models of macromolecular structures derived from X-ray diffraction data have their limitations, which need to be critically evaluated and well understood for structure-based drug discovery.
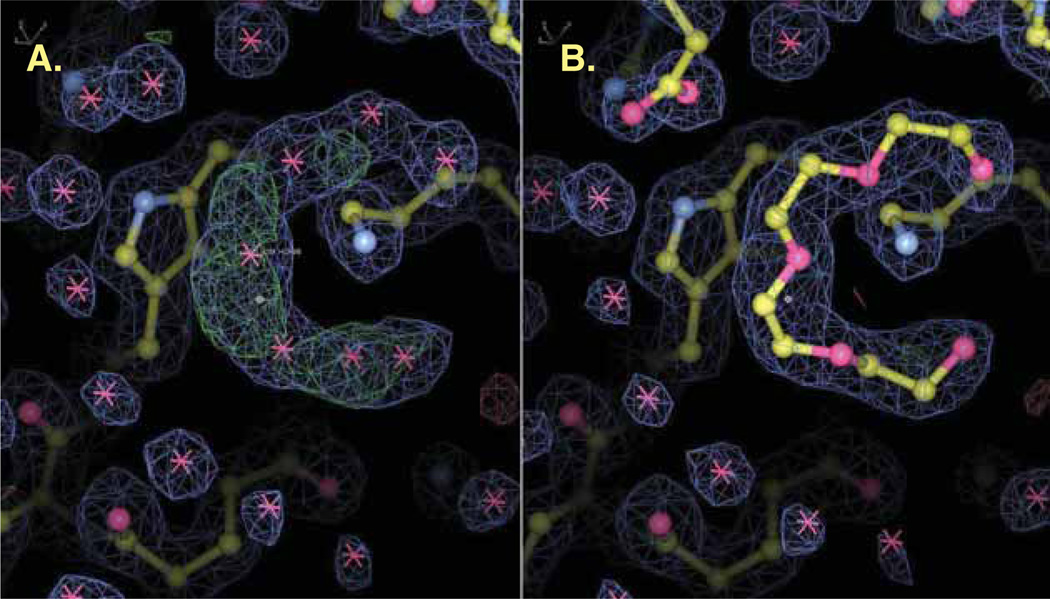
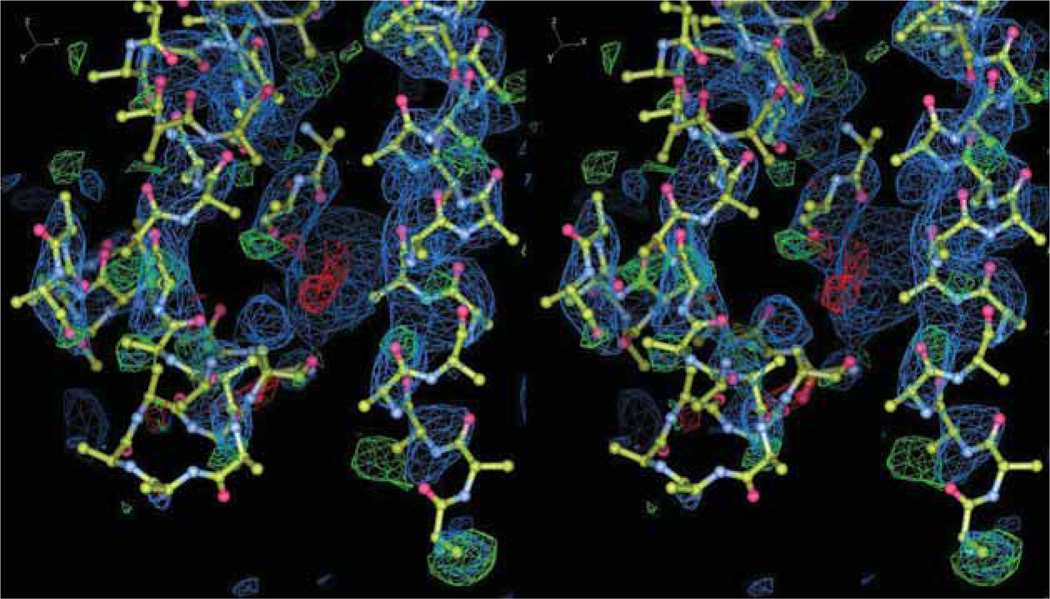
This review describes how the validity, accuracy and precision of a protein or nucleic acid structure determined by X-ray crystallography can be evaluated from three different perspectives: i) the nature of the diffraction experiment; ii) the interpretation of an electron density map; and iii) the interpretation of the structural model in terms of function and mechanism. The strategies to optimally exploit a macromolecular structure are also discussed in the context of 'Big Data' analysis, biochemical experimental design and structure-based drug discovery.
Although X-ray crystallography is one of the most detailed 'microscopes' available today for examining macromolecular structures, the authors would like to re-emphasize that such structures are only simplified models of the target macromolecules. The authors also wish to reinforce the idea that a structure should not be thought of as a set of precise coordinates but rather as a framework for generating hypotheses to be explored. Numerous biochemical and biophysical experiments, including new diffraction experiments, can and should be performed to verify or falsify these hypotheses. X-ray crystallography will find its future application in drug discovery by the development of specific tools that would allow realistic interpretation of the outcome coordinates and/or support testing of these hypotheses.
X射线晶体学在基于结构的药物设计(SBDD)中发挥着重要作用,在各个阶段对靶标大分子及大分子-配体复合物晶体结构进行准确分析至关重要。然而,尽管在改进结构生物学方法方面取得了显著进展,尤其是在X射线晶体学领域,但计算方法(如计算机辅助高通量筛选)的相应进展仍有待实现。晶体结构可能被过度解读,从而使假设和后续实验产生偏差。与任何实验科学一样,从X射线衍射数据推导得到的大分子结构模型存在局限性,在基于结构的药物发现中需要对这些局限性进行严格评估并充分理解。
本综述描述了如何从三个不同角度评估通过X射线晶体学确定的蛋白质或核酸结构的有效性、准确性和精确性:i)衍射实验的性质;ii)电子密度图的解读;iii)根据功能和机制对结构模型的解读。还在“大数据”分析、生化实验设计和基于结构药物发现的背景下讨论了最佳利用大分子结构的策略。
尽管X射线晶体学是当今用于研究大分子结构的最详细的“显微镜”之一,但作者想再次强调,此类结构只是靶标大分子的简化模型。作者还希望强化这样一种观点,即不应将结构视为一组精确的坐标,而应将其视为生成有待探索假设的框架。可以且应该进行大量生化和生物物理实验,包括新的衍射实验,以验证或证伪这些假设。通过开发能够对结果坐标进行实际解读和/或支持对这些假设进行测试的特定工具,X射线晶体学将在药物发现中找到其未来应用。