Sabarinathan Radhakrishnan, Wenzel Anne, Novotny Peter, Tang Xiaojia, Kalari Krishna R, Gorodkin Jan
Center for non-coding RNA in Technology and Health, Section for Animal Genetics, Bioinformatics and Breeding, IKVH, University of Copenhagen, Frederiksberg, Denmark.
Bioinformatics Centre, Department of Biology and Biotech Research and Innovation Centre, University of Copenhagen, Copenhagen, Denmark.
PLoS One. 2014 Jan 8;9(1):e82699. doi: 10.1371/journal.pone.0082699. eCollection 2014.
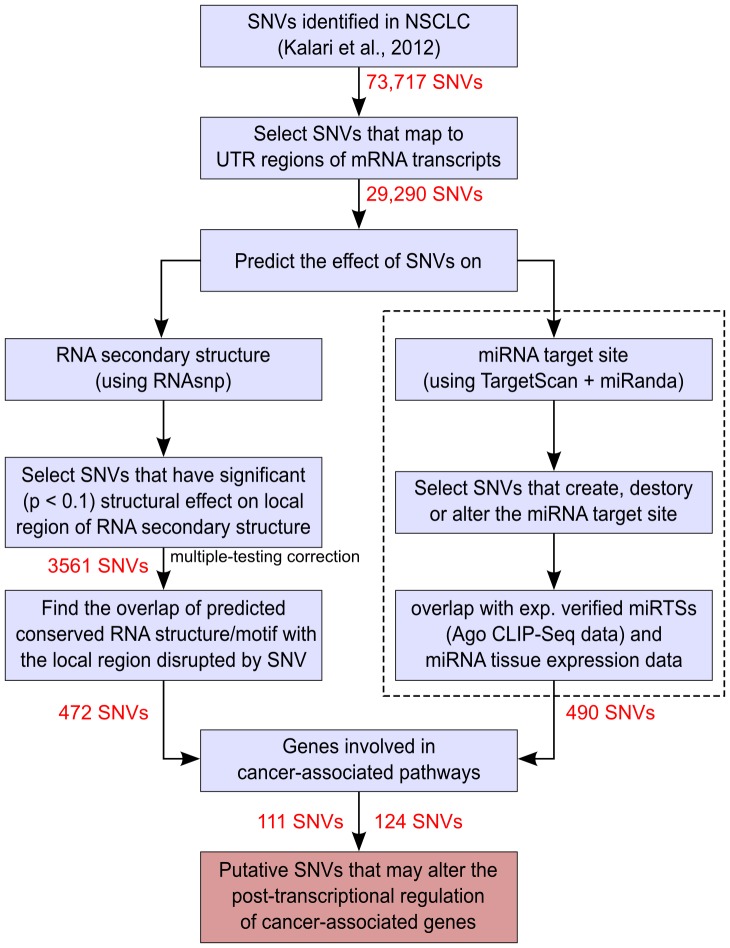
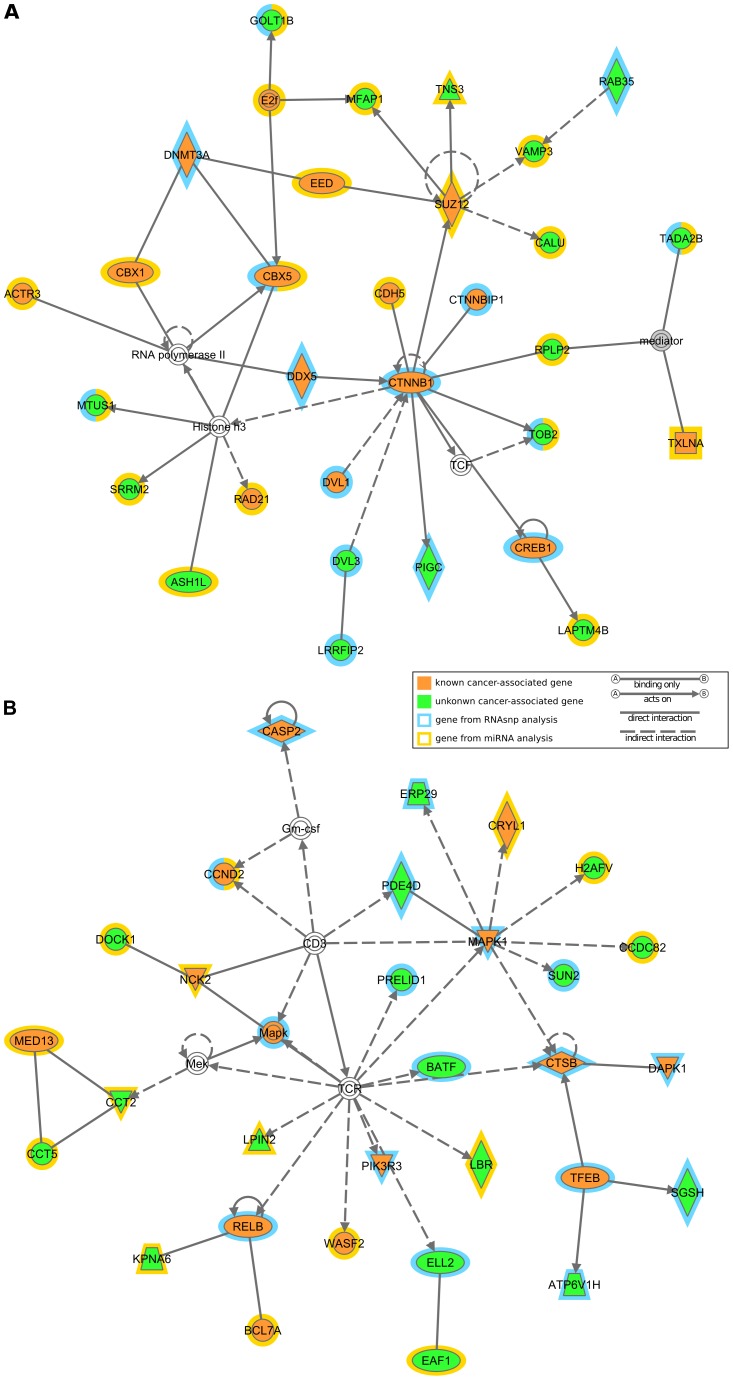
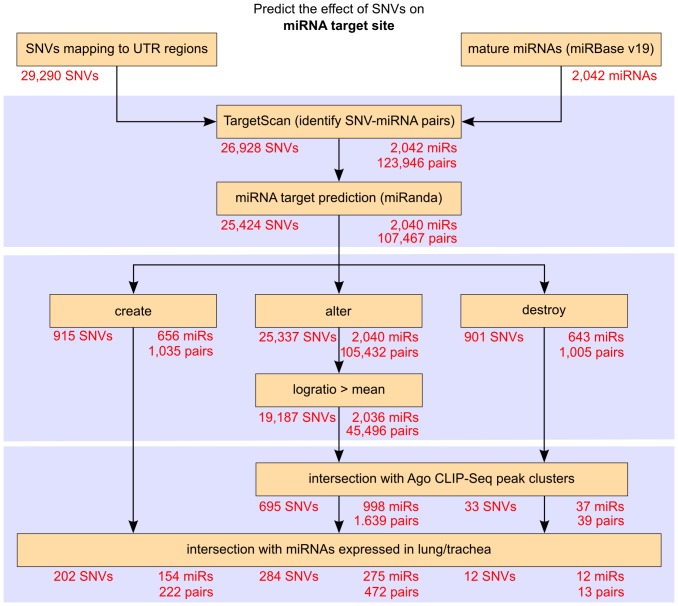
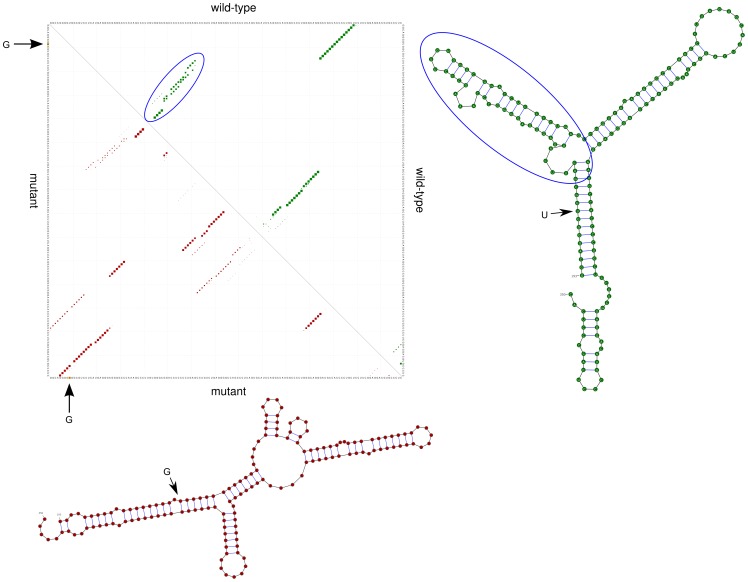
Traditional mutation assessment methods generally focus on predicting disruptive changes in protein-coding regions rather than non-coding regulatory regions like untranslated regions (UTRs) of mRNAs. The UTRs, however, are known to have many sequence and structural motifs that can regulate translational and transcriptional efficiency and stability of mRNAs through interaction with RNA-binding proteins and other non-coding RNAs like microRNAs (miRNAs). In a recent study, transcriptomes of tumor cells harboring mutant and wild-type KRAS (V-Ki-ras2 Kirsten rat sarcoma viral oncogene homolog) genes in patients with non-small cell lung cancer (NSCLC) have been sequenced to identify single nucleotide variations (SNVs). About 40% of the total SNVs (73,717) identified were mapped to UTRs, but omitted in the previous analysis. To meet this obvious demand for analysis of the UTRs, we designed a comprehensive pipeline to predict the effect of SNVs on two major regulatory elements, secondary structure and miRNA target sites. Out of 29,290 SNVs in 6462 genes, we predict 472 SNVs (in 408 genes) affecting local RNA secondary structure, 490 SNVs (in 447 genes) affecting miRNA target sites and 48 that do both. Together these disruptive SNVs were present in 803 different genes, out of which 188 (23.4%) were previously known to be cancer-associated. Notably, this ratio is significantly higher (one-sided Fisher's exact test p-value = 0.032) than the ratio (20.8%) of known cancer-associated genes (n = 1347) in our initial data set (n = 6462). Network analysis shows that the genes harboring disruptive SNVs were involved in molecular mechanisms of cancer, and the signaling pathways of LPS-stimulated MAPK, IL-6, iNOS, EIF2 and mTOR. In conclusion, we have found hundreds of SNVs which are highly disruptive with respect to changes in the secondary structure and miRNA target sites within UTRs. These changes hold the potential to alter the expression of known cancer genes or genes linked to cancer-associated pathways.
传统的突变评估方法通常侧重于预测蛋白质编码区域的破坏性变化,而非像mRNA的非翻译区(UTRs)这样的非编码调控区域。然而,已知UTR具有许多序列和结构基序,这些基序可通过与RNA结合蛋白以及其他非编码RNA(如微小RNA,miRNAs)相互作用来调节mRNA的翻译、转录效率和稳定性。在最近一项研究中,对非小细胞肺癌(NSCLC)患者中携带突变型和野生型KRAS(V-Ki-ras2 Kirsten大鼠肉瘤病毒癌基因同源物)基因的肿瘤细胞转录组进行了测序,以鉴定单核苷酸变异(SNVs)。所鉴定的全部SNVs(73,717个)中约40%定位于UTR,但在先前的分析中被遗漏。为满足对UTR分析的这一明显需求,我们设计了一套综合流程来预测SNVs对两个主要调控元件(二级结构和miRNA靶位点)的影响。在6462个基因中的29,290个SNVs中,我们预测有472个SNVs(存在于408个基因中)影响局部RNA二级结构,490个SNVs(存在于447个基因中)影响miRNA靶位点,还有48个同时影响二者。这些具有破坏性的SNVs共存在于803个不同基因中,其中188个(23.4%)先前已知与癌症相关。值得注意的是,这一比例显著高于我们初始数据集(n = 6462)中已知癌症相关基因(n = 1347)的比例(20.8%)(单侧Fisher精确检验p值 = 0.032)。网络分析表明,携带破坏性SNVs的基因参与了癌症的分子机制以及LPS刺激的MAPK、IL-6、iNOS、EIF2和mTOR信号通路。总之,我们发现了数百个对UTR内二级结构和miRNA靶位点变化具有高度破坏性的SNVs。这些变化有可能改变已知癌症基因或与癌症相关通路相关基因的表达。