Tang Wai Ho, Stitham Jeremiah, Jin Yu, Liu Renjing, Lee Seung Hee, Du Jing, Atteya Gourg, Gleim Scott, Spollett Geralyn, Martin Kathleen, Hwa John
Yale Cardiovascular Research Center, Section of Cardiovascular Medicine (W.H.T., J.S., Y.J., R.L., S.H.L., J.D., G.A., S.G., K.M., J.H.) and Section of Endocrinology and Metabolism, Department of Internal Medicine (G.S.), Yale University School of Medicine, New Haven, CT.
Circulation. 2014 Apr 15;129(15):1598-609. doi: 10.1161/CIRCULATIONAHA.113.005224. Epub 2014 Jan 28.
Platelet abnormalities are well-recognized complications of diabetes mellitus. Mitochondria play a central role in platelet metabolism and activation. Mitochondrial dysfunction is evident in diabetes mellitus. The molecular pathway for hyperglycemia-induced mitochondrial dysfunction in platelets in diabetes mellitus is unknown.
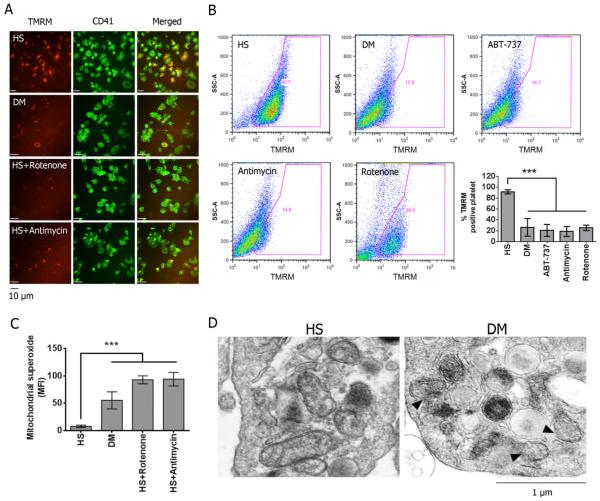
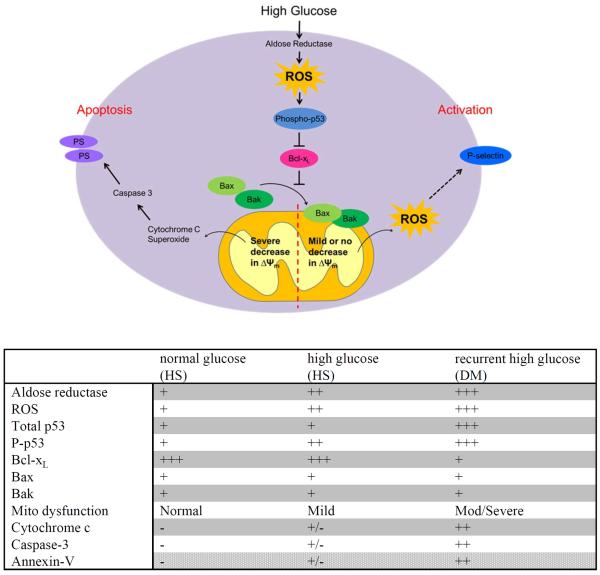
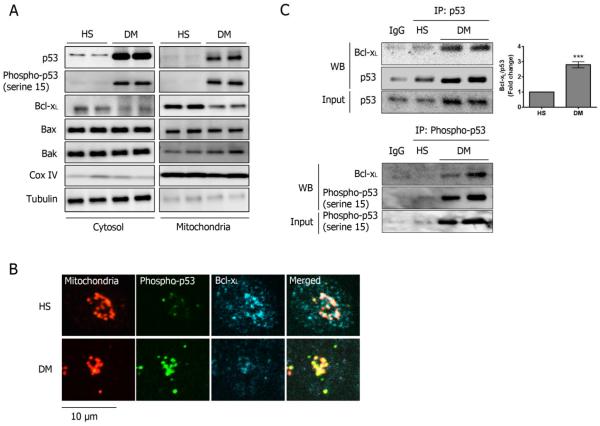
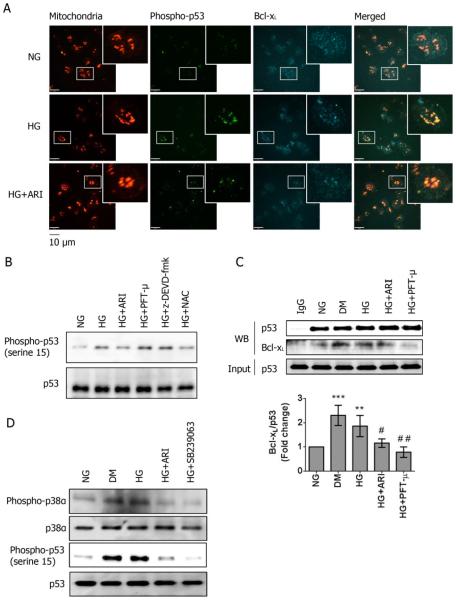
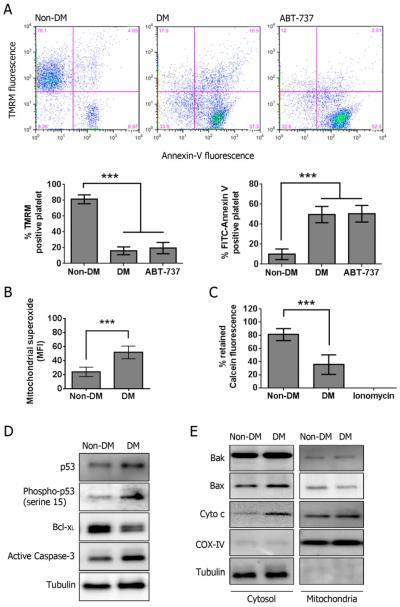
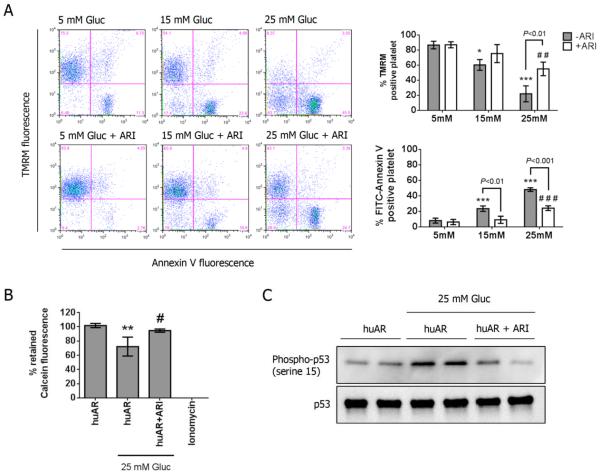
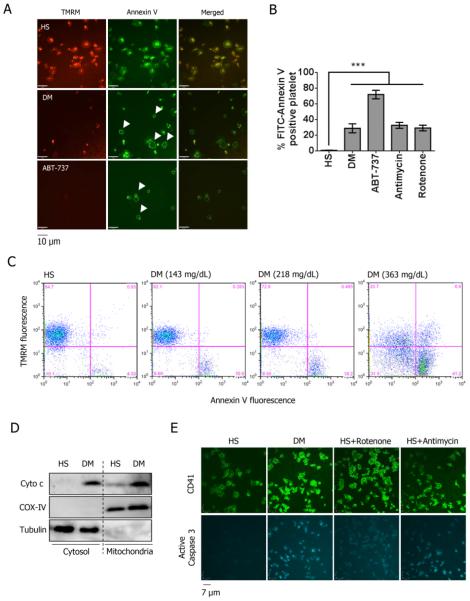
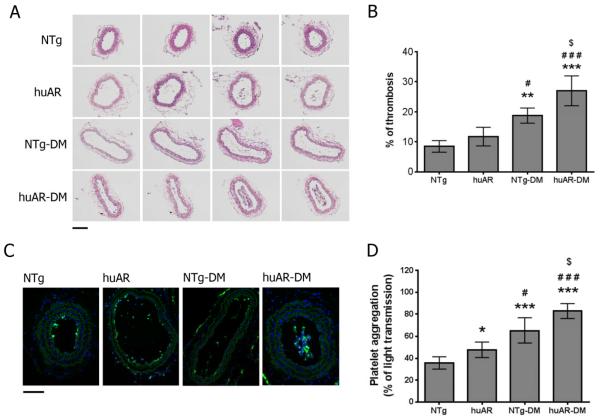
Using both human and humanized mouse models, we report that hyperglycemia-induced aldose reductase activation and subsequent reactive oxygen species production lead to increased p53 phosphorylation (Ser15), which promotes mitochondrial dysfunction, damage, and rupture by sequestration of the antiapoptotic protein Bcl-xL. In a glucose dose-dependent manner, severe mitochondrial damage leads to loss of mitochondrial membrane potential and platelet apoptosis (cytochrome c release, caspase 3 activation, and phosphatidylserine exposure). Although platelet hyperactivation, mitochondrial dysfunction, aldose reductase activation, reactive oxygen species production, and p53 phosphorylation are all induced by hyperglycemia, we demonstrate that platelet apoptosis and hyperactivation are 2 distinct states that depend on the severity of the hyperglycemia and mitochondrial damage. Combined, both lead to increased thrombus formation in a mouse blood stasis model.
Aldose reductase contributes to diabetes-mediated mitochondrial dysfunction and damage through the activation of p53. The degree of mitochondrial dysfunction and damage determines whether hyperactivity (mild damage) or apoptosis (severe damage) will ensue. These signaling components provide novel therapeutic targets for thrombotic complications in diabetes mellitus.
血小板异常是糖尿病公认的并发症。线粒体在血小板代谢和激活中起核心作用。线粒体功能障碍在糖尿病中很明显。糖尿病中高血糖诱导血小板线粒体功能障碍的分子途径尚不清楚。
使用人类和人源化小鼠模型,我们报告高血糖诱导的醛糖还原酶激活及随后的活性氧生成导致p53磷酸化(Ser15)增加,这通过隔离抗凋亡蛋白Bcl-xL促进线粒体功能障碍、损伤和破裂。以葡萄糖剂量依赖的方式,严重的线粒体损伤导致线粒体膜电位丧失和血小板凋亡(细胞色素c释放、半胱天冬酶3激活和磷脂酰丝氨酸暴露)。虽然血小板过度激活、线粒体功能障碍、醛糖还原酶激活、活性氧生成和p53磷酸化均由高血糖诱导,但我们证明血小板凋亡和过度激活是两种不同的状态,取决于高血糖和线粒体损伤的严重程度。两者共同导致小鼠血瘀模型中血栓形成增加。
醛糖还原酶通过激活p53导致糖尿病介导的线粒体功能障碍和损伤。线粒体功能障碍和损伤的程度决定了随后是过度活跃(轻度损伤)还是凋亡(严重损伤)。这些信号成分提供了糖尿病血栓性并发症的新治疗靶点。