Matsumoto Ayumi, Mizuno Makoto, Hamada Nanako, Nozaki Yasuyuki, Jimbo Eriko F, Momoi Mariko Y, Nagata Koh-ichi, Yamagata Takanori
Department of Pediatrics, Jichi Medical University, Tochigi, Japan.
Department of Molecular Neurobiology, Institute for Developmental Research, Aichi Human Service Center, Kasugai, Japan.
PLoS One. 2014 Mar 21;9(3):e92695. doi: 10.1371/journal.pone.0092695. eCollection 2014.
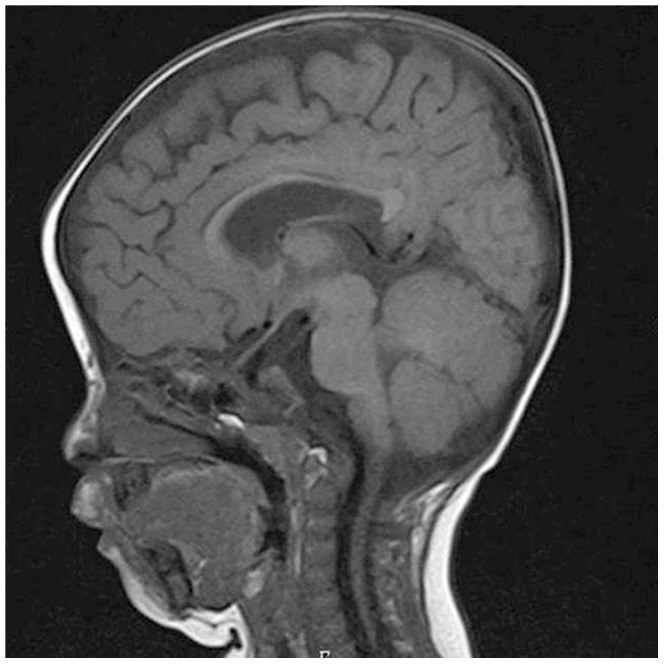
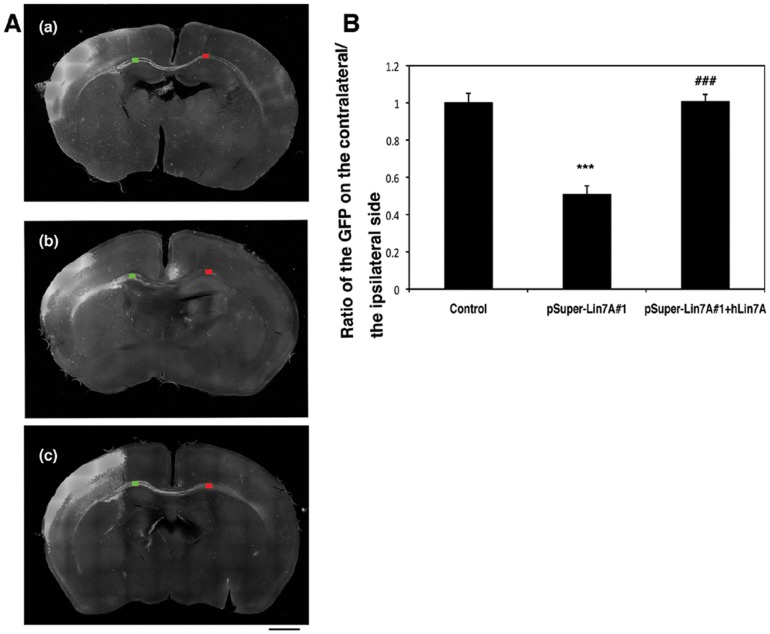
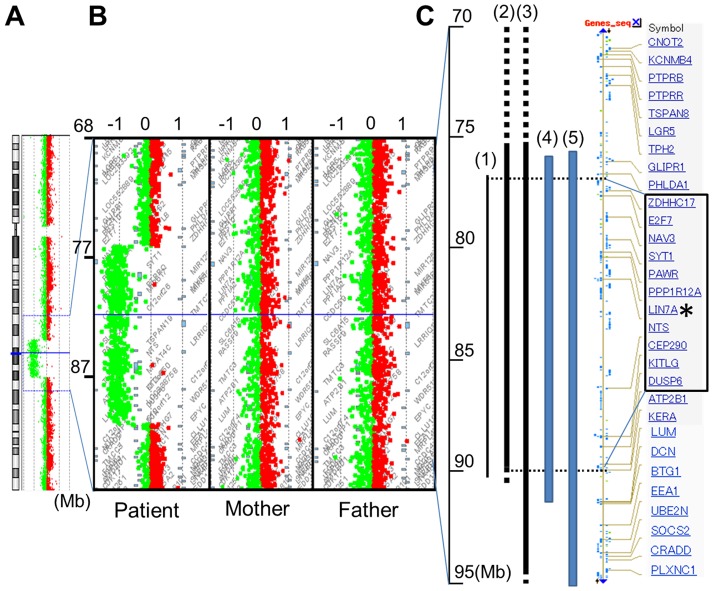
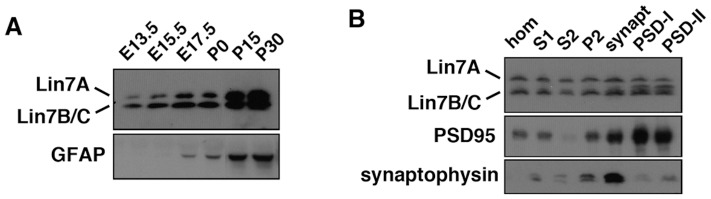
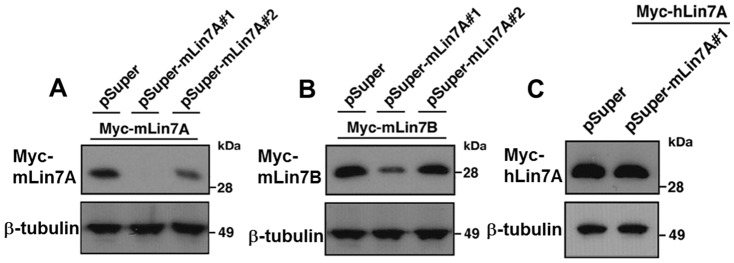
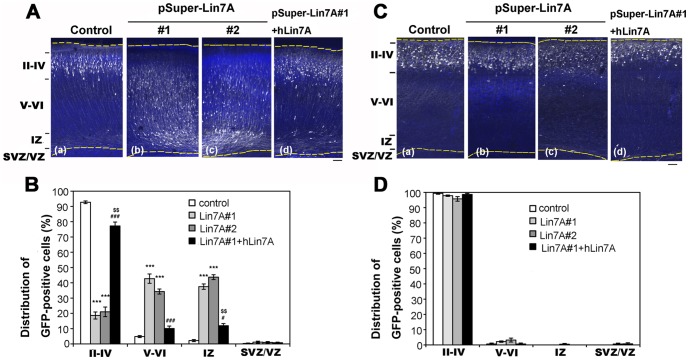
Interstitial deletion of 12q21 has been reported in four cases, which share several common clinical features, including intellectual disability (ID), low-set ears, and minor cardiac abnormalities. Comparative genomic hybridization (CGH) analysis using the Agilent Human Genome CGH 180K array was performed with the genomic DNA from a two-year-old Japanese boy with these symptoms, as well as hypoplasia of the corpus callosum. Consequently, a 14 Mb deletion at 12q21.2-q21.33 (nt. 77 203 574-91 264 613 bp), which includes 72 genes, was detected. Of these, we focused on LIN7A, which encodes a scaffold protein that is important for synaptic function, as a possible responsible gene for ID, and we analyzed its role in cerebral cortex development. Western blotting analyses revealed that Lin-7A is expressed on embryonic day (E) 13.5, and gradually increases in the mouse brain during the embryonic stage. Biochemical fractionation resulted in the enrichment of Lin-7A in the presynaptic fraction. Suppression of Lin-7A expression by RNAi, using in utero electroporation on E14.5, delayed neuronal migration on postnatal day (P) 2, and Lin-7A-deficient neurons remained in the lower zone of the cortical plate and the intermediate zone. In addition, when Lin-7A was silenced in cortical neurons in one hemisphere, axonal growth in the contralateral hemisphere was delayed; development of these neurons was disrupted such that one half did not extend into the contralateral hemisphere after leaving the corpus callosum. Taken together, LIN7A is a candidate gene responsible for 12q21-deletion syndrome, and abnormal neuronal migration and interhemispheric axon development may contribute to ID and corpus callosum hypoplasia, respectively.
已有报道称,4例患者存在12q21间质缺失,他们具有一些共同的临床特征,包括智力残疾(ID)、低位耳和轻微心脏异常。对一名患有这些症状以及胼胝体发育不全的两岁日本男孩的基因组DNA,使用安捷伦人类基因组CGH 180K芯片进行了比较基因组杂交(CGH)分析。结果检测到12q21.2-q21.33(nt. 77 203 574-91 264 613 bp)处有一个14 Mb的缺失,其中包含72个基因。其中,我们将重点关注LIN7A,它编码一种对突触功能很重要的支架蛋白,可能是导致ID的致病基因,并分析了它在大脑皮层发育中的作用。蛋白质免疫印迹分析显示,Lin-7A在胚胎第13.5天(E13.5)表达,并在胚胎期小鼠大脑中逐渐增加。生化分级分离导致Lin-7A在前突触部分富集。在E14.5进行子宫内电穿孔,通过RNAi抑制Lin-7A的表达,导致出生后第2天(P2)神经元迁移延迟,Lin-7A缺陷神经元仍留在皮质板的下部区域和中间区域。此外,当在一个半球的皮质神经元中沉默Lin-7A时,对侧半球的轴突生长会延迟;这些神经元的发育受到破坏,以至于一半神经元在离开胼胝体后没有延伸到对侧半球。综上所述,LIN7A是导致12q21缺失综合征的候选基因,异常的神经元迁移和半球间轴突发育可能分别导致ID和胼胝体发育不全。