Santpere Gabriel, Darre Fleur, Blanco Soledad, Alcami Antonio, Villoslada Pablo, Mar Albà M, Navarro Arcadi
Institut de Biologia Evolutiva (Universitat Pompeu Fabra - CSIC), Barcelona, Spain.
Genome Biol Evol. 2014 Apr;6(4):846-60. doi: 10.1093/gbe/evu054.
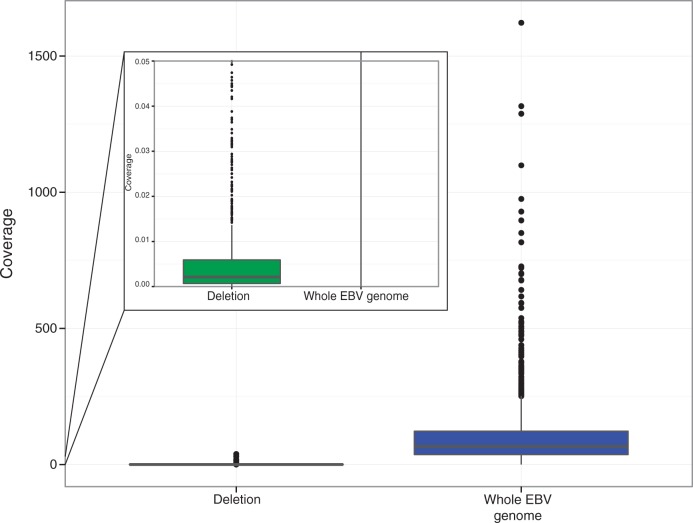
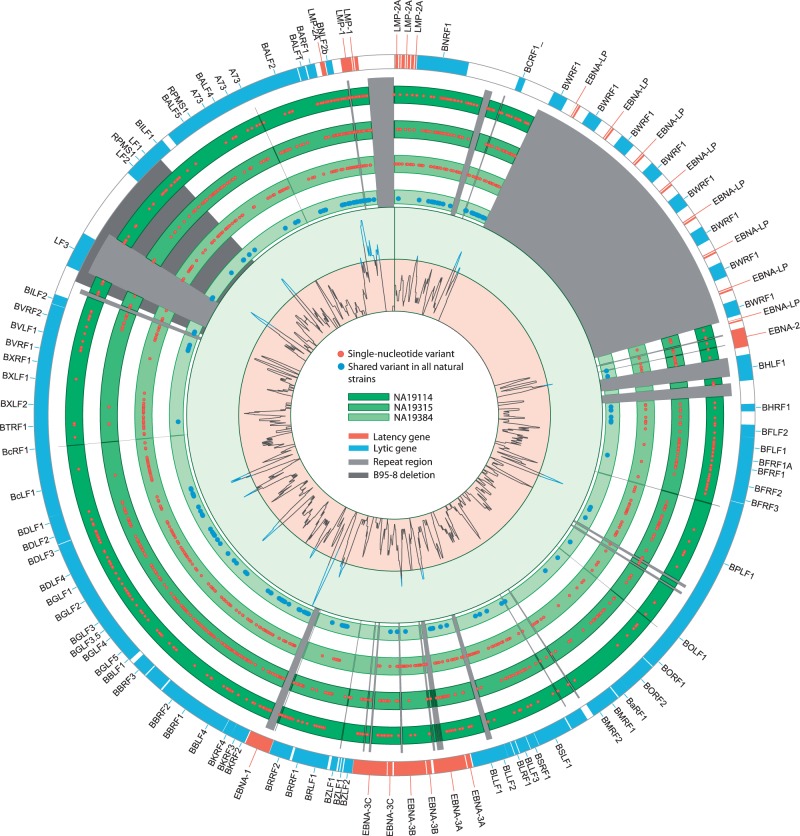
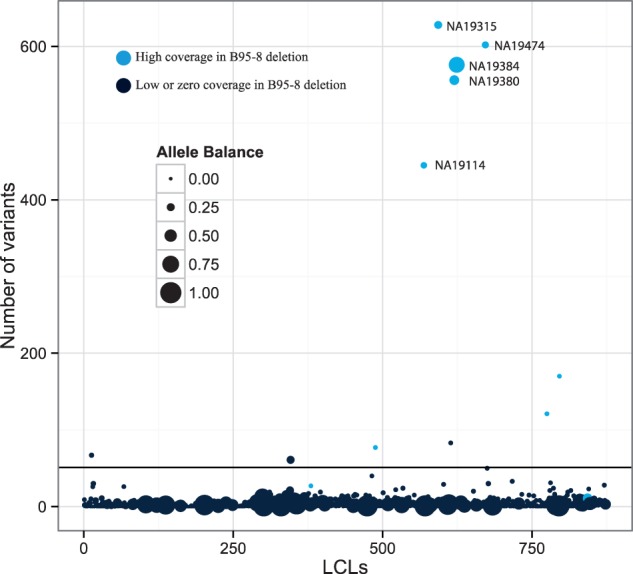
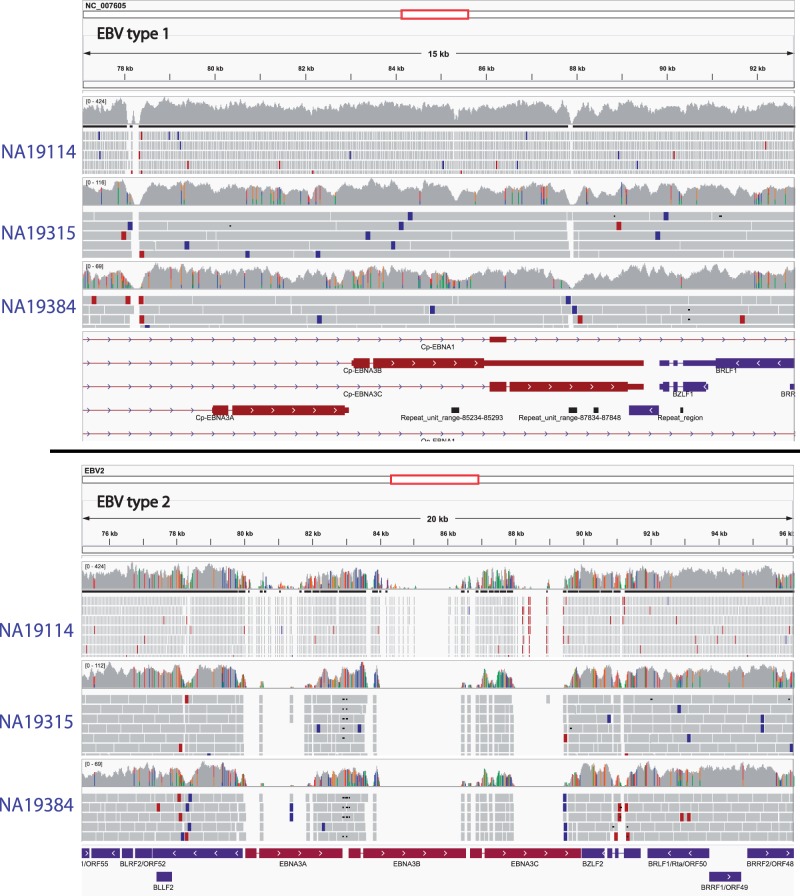
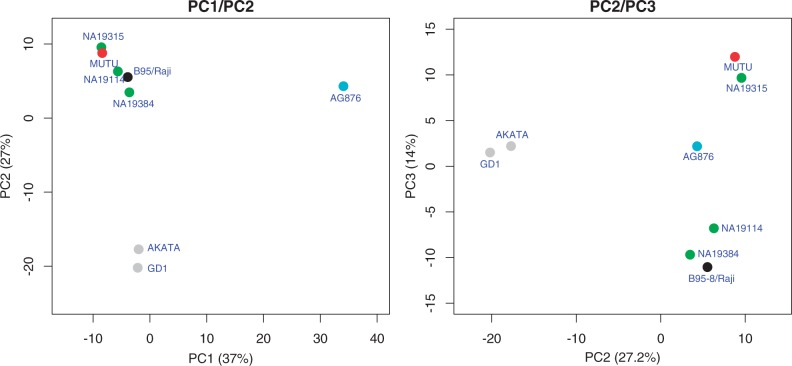
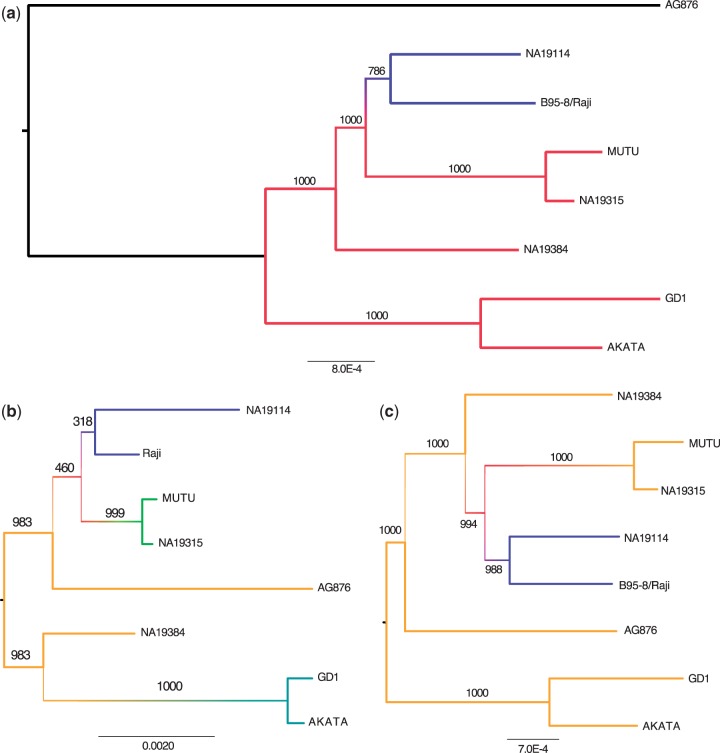
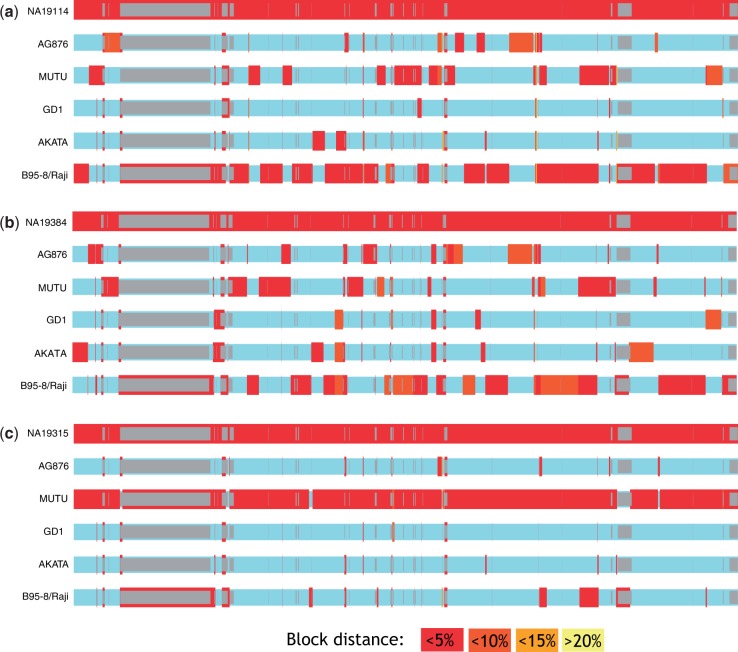
Most people in the world (∼90%) are infected by the Epstein-Barr virus (EBV), which establishes itself permanently in B cells. Infection by EBV is related to a number of diseases including infectious mononucleosis, multiple sclerosis, and different types of cancer. So far, only seven complete EBV strains have been described, all of them coming from donors presenting EBV-related diseases. To perform a detailed comparative genomic analysis of EBV including, for the first time, EBV strains derived from healthy individuals, we reconstructed EBV sequences infecting lymphoblastoid cell lines (LCLs) from the 1000 Genomes Project. As strain B95-8 was used to transform B cells to obtain LCLs, it is always present, but a specific deletion in its genome sets it apart from natural EBV strains. After studying hundreds of individuals, we determined the presence of natural EBV in at least 10 of them and obtained a set of variants specific to wild-type EBV. By mapping the natural EBV reads into the EBV reference genome (NC007605), we constructed nearly complete wild-type viral genomes from three individuals. Adding them to the five disease-derived EBV genomic sequences available in the literature, we performed an in-depth comparative genomic analysis. We found that latency genes harbor more nucleotide diversity than lytic genes and that six out of nine latency-related genes, as well as other genes involved in viral attachment and entry into host cells, packaging, and the capsid, present the molecular signature of accelerated protein evolution rates, suggesting rapid host-parasite coevolution.
世界上大多数人(约90%)感染过爱泼斯坦-巴尔病毒(EBV),该病毒会在B细胞中永久存在。EBV感染与多种疾病有关,包括传染性单核细胞增多症、多发性硬化症和不同类型的癌症。到目前为止,仅描述了七种完整的EBV毒株,所有这些毒株均来自患有EBV相关疾病的供体。为了首次对EBV进行详细的比较基因组分析,包括来自健康个体的EBV毒株,我们从千人基因组计划中重建了感染淋巴母细胞系(LCLs)的EBV序列。由于使用B95 - 8毒株来转化B细胞以获得LCLs,它总是存在,但其基因组中的特定缺失使其与天然EBV毒株区分开来。在研究了数百名个体后,我们确定其中至少10人存在天然EBV,并获得了一组野生型EBV特有的变体。通过将天然EBV读数映射到EBV参考基因组(NC007605)中,我们从三个个体构建了近乎完整的野生型病毒基因组。将它们添加到文献中现有的五个疾病来源的EBV基因组序列中,我们进行了深入的比较基因组分析。我们发现潜伏基因比裂解基因具有更多的核苷酸多样性,并且九个潜伏相关基因中的六个,以及其他参与病毒附着、进入宿主细胞、包装和衣壳的基因,呈现出蛋白质进化速率加快的分子特征,表明宿主与寄生虫之间存在快速的共同进化。