Wright David W, Hall Benjamin A, Kenway Owain A, Jha Shantenu, Coveney Peter V
Centre for Computational Science, Department of Chemistry, University College London , London WC1H 0AJ, United Kingdom.
Electrical and Computer Engineering, Rutgers University , Piscataway, New Jersey 08854, United States.
J Chem Theory Comput. 2014 Mar 11;10(3):1228-1241. doi: 10.1021/ct4007037. Epub 2014 Jan 27.
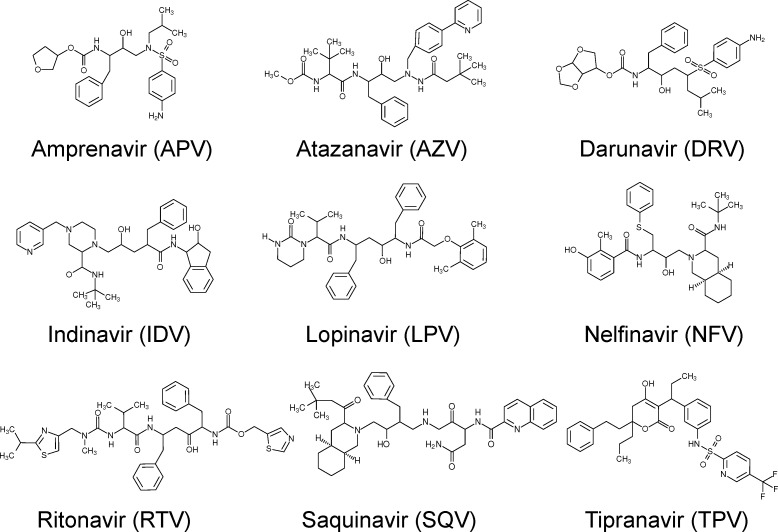
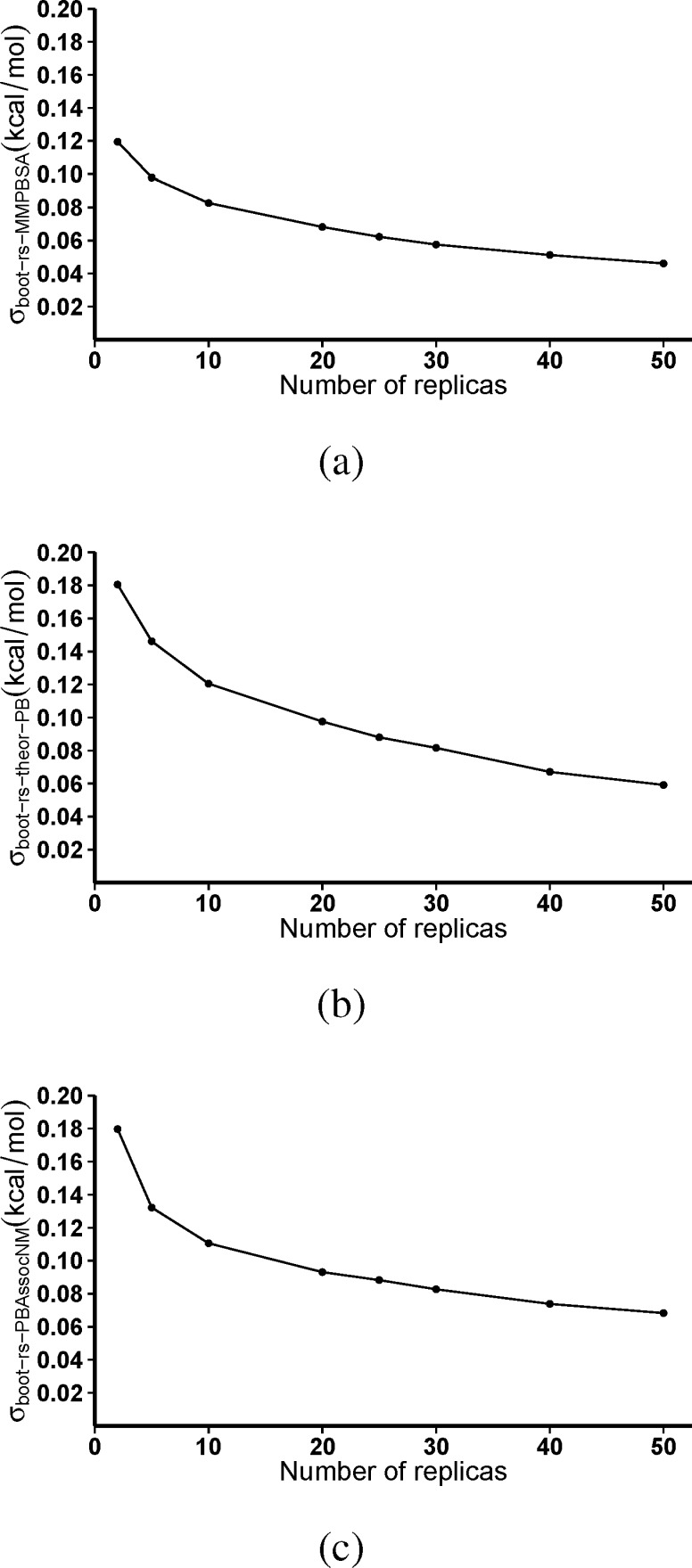
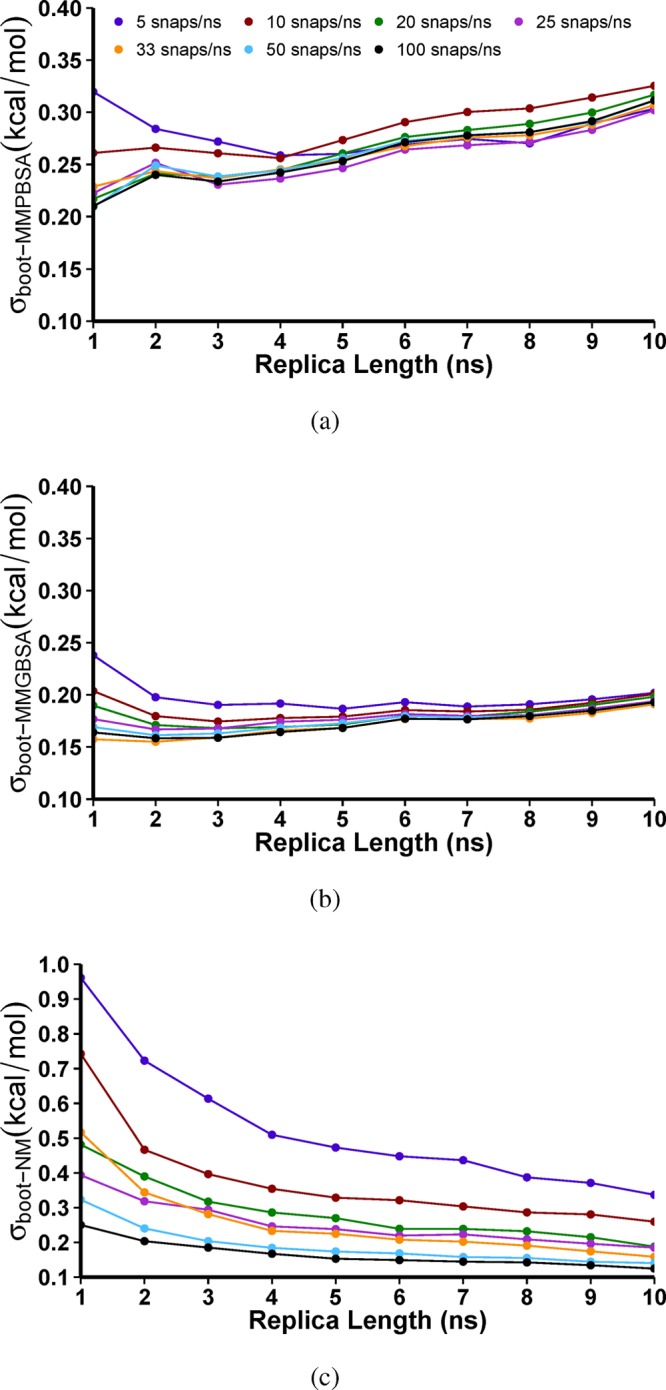
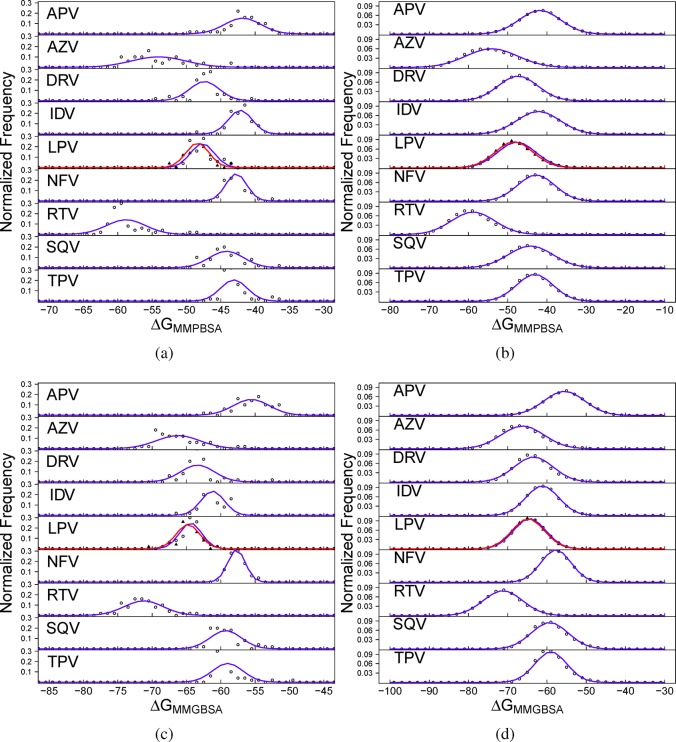
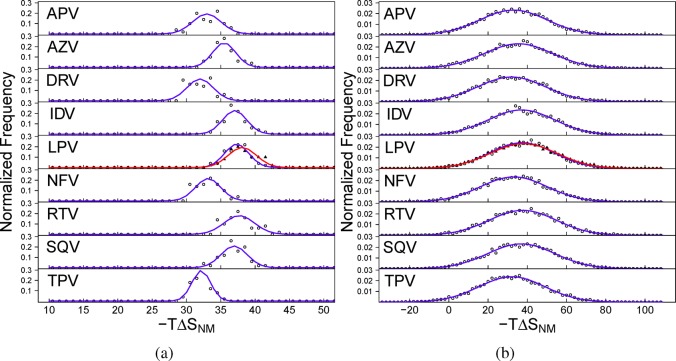
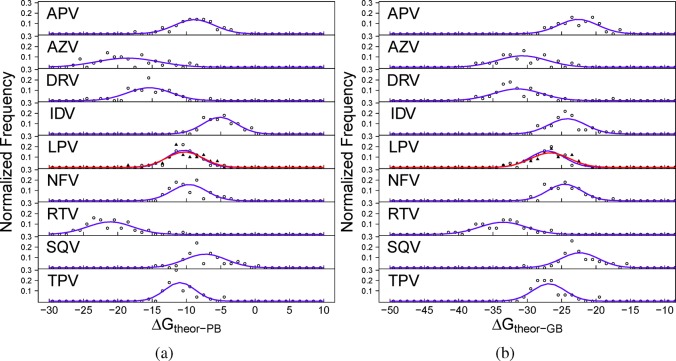
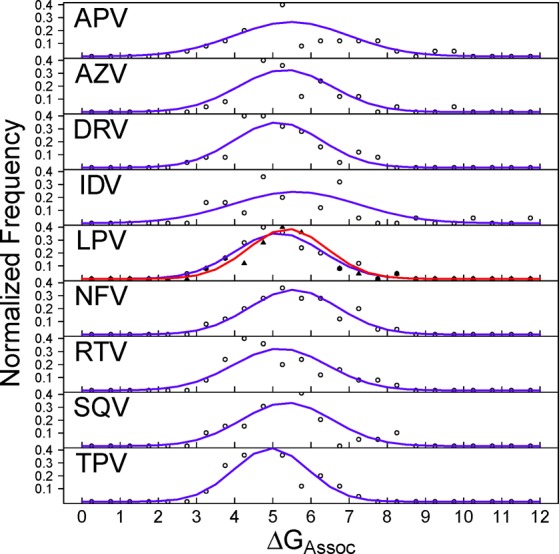
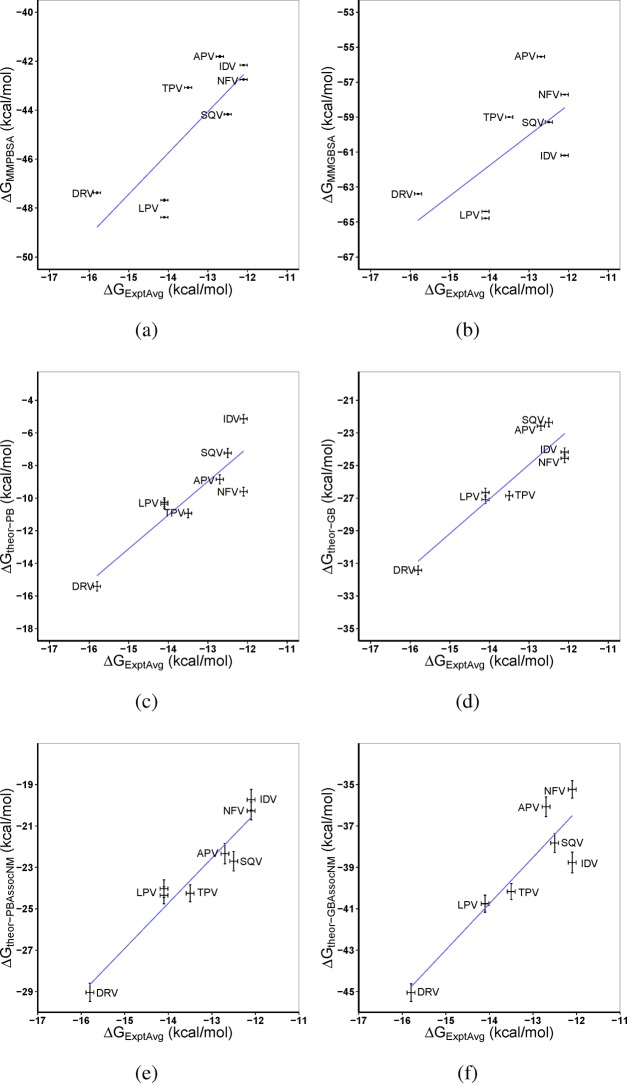
The use of molecular simulation to estimate the strength of macromolecular binding free energies is becoming increasingly widespread, with goals ranging from lead optimization and enrichment in drug discovery to personalizing or stratifying treatment regimes. In order to realize the potential of such approaches to predict new results, not merely to explain previous experimental findings, it is necessary that the methods used are reliable and accurate, and that their limitations are thoroughly understood. However, the computational cost of atomistic simulation techniques such as molecular dynamics (MD) has meant that until recently little work has focused on validating and verifying the available free energy methodologies, with the consequence that many of the results published in the literature are not reproducible. Here, we present a detailed analysis of two of the most popular approximate methods for calculating binding free energies from molecular simulations, molecular mechanics Poisson-Boltzmann surface area (MMPBSA) and molecular mechanics generalized Born surface area (MMGBSA), applied to the nine FDA-approved HIV-1 protease inhibitors. Our results show that the values obtained from replica simulations of the same protease-drug complex, differing only in initially assigned atom velocities, can vary by as much as 10 kcal mol, which is greater than the difference between the best and worst binding inhibitors under investigation. Despite this, analysis of ensembles of simulations producing 50 trajectories of 4 ns duration leads to well converged free energy estimates. For seven inhibitors, we find that with correctly converged normal mode estimates of the configurational entropy, we can correctly distinguish inhibitors in agreement with experimental data for both the MMPBSA and MMGBSA methods and thus have the ability to rank the efficacy of binding of this selection of drugs to the protease (no account is made for free energy penalties associated with protein distortion leading to the over estimation of the binding strength of the two largest inhibitors ritonavir and atazanavir). We obtain improved rankings and estimates of the relative binding strengths of the drugs by using a novel combination of MMPBSA/MMGBSA with normal mode entropy estimates and the free energy of association calculated directly from simulation trajectories. Our work provides a thorough assessment of what is required to produce converged and hence reliable free energies for protein-ligand binding.
利用分子模拟来估算大分子结合自由能的强度正变得越来越普遍,其目标涵盖从药物发现中的先导优化和富集到个性化或分层治疗方案等各个方面。为了实现此类方法预测新结果的潜力,而不仅仅是解释先前的实验发现,所使用的方法必须可靠且准确,并且其局限性也必须被透彻理解。然而,诸如分子动力学(MD)等原子模拟技术的计算成本意味着,直到最近,很少有工作专注于验证和核实现有的自由能方法,结果是文献中发表的许多结果都无法重现。在此,我们对两种最流行的从分子模拟计算结合自由能的近似方法——分子力学泊松-玻尔兹曼表面积(MMPBSA)和分子力学广义玻恩表面积(MMGBSA)进行了详细分析,这些方法应用于九种FDA批准的HIV-1蛋白酶抑制剂。我们的结果表明,从相同蛋白酶-药物复合物的副本模拟中获得的值,仅在初始分配的原子速度上有所不同,其变化幅度可达10千卡/摩尔,这大于所研究的最佳和最差结合抑制剂之间的差异。尽管如此,对产生50条持续时间为4纳秒轨迹的模拟系综进行分析,可得到收敛良好的自由能估计值。对于七种抑制剂,我们发现通过正确收敛的构型熵的简正模式估计,我们能够正确区分抑制剂,这与MMPBSA和MMGBSA方法的实验数据一致,因此有能力对这组药物与蛋白酶的结合效力进行排序(未考虑与蛋白质变形相关的自由能惩罚,这导致对两种最大的抑制剂利托那韦和阿扎那韦的结合强度估计过高)。通过将MMPBSA/MMGBSA与简正模式熵估计以及直接从模拟轨迹计算的缔合自由能进行新颖组合,我们获得了改进的药物相对结合强度排名和估计值。我们的工作对产生收敛且因此可靠的蛋白质-配体结合自由能所需的条件进行了全面评估。