Bio Systems Analysis Group, Institute of Computer Science, Jena Centre for Bioinformatics and Friedrich Schiller University Jena, Ernst-Abbe-Platz 2, D-0007743 Jena, Germany.
Cells. 2013 Jul 2;2(3):506-44. doi: 10.3390/cells2030506.
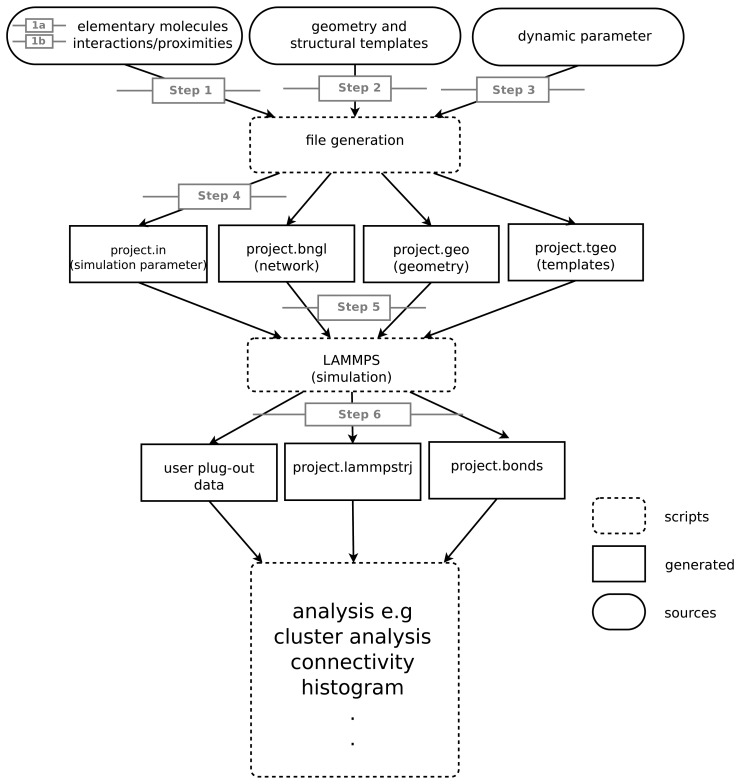
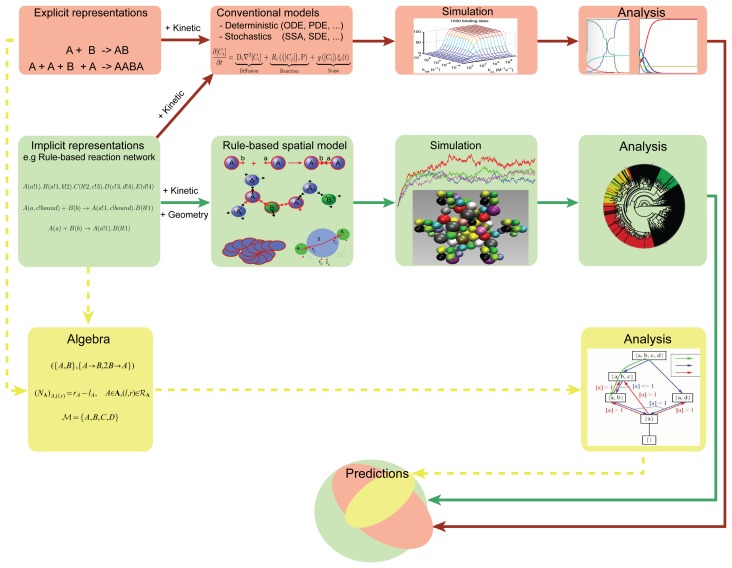
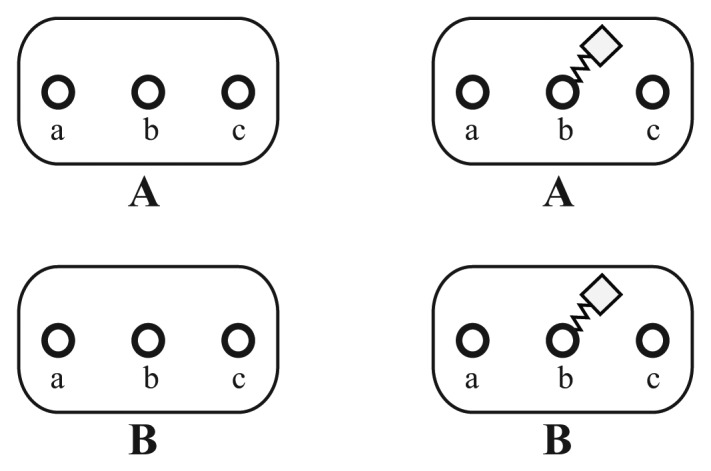
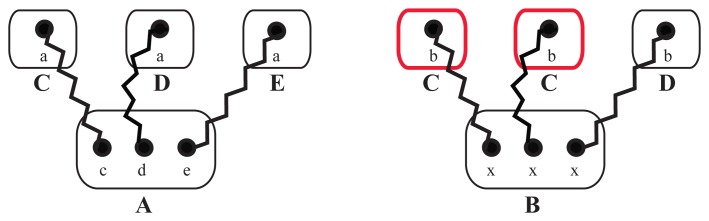
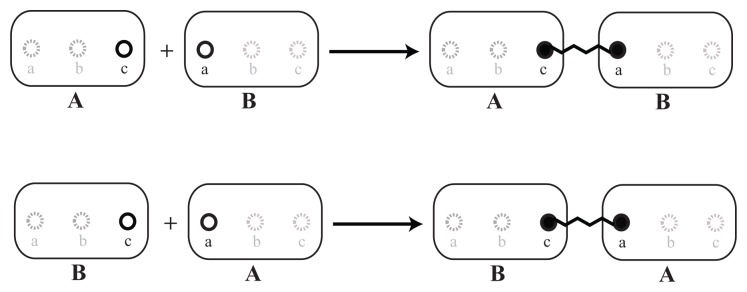
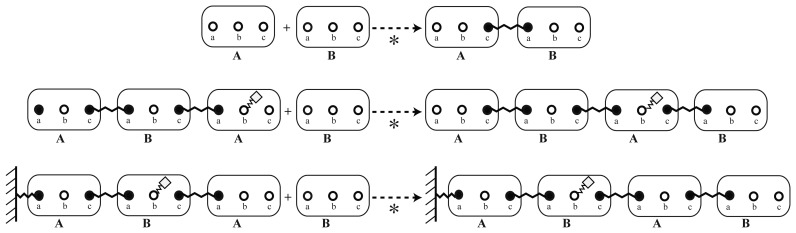
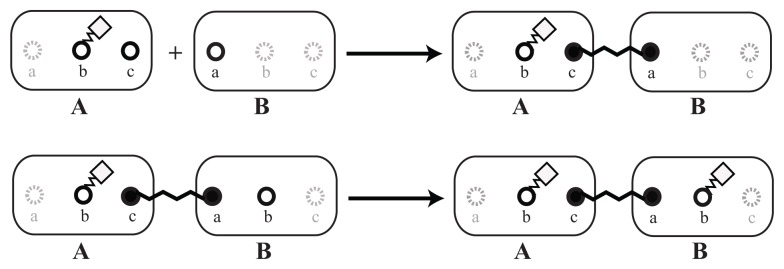
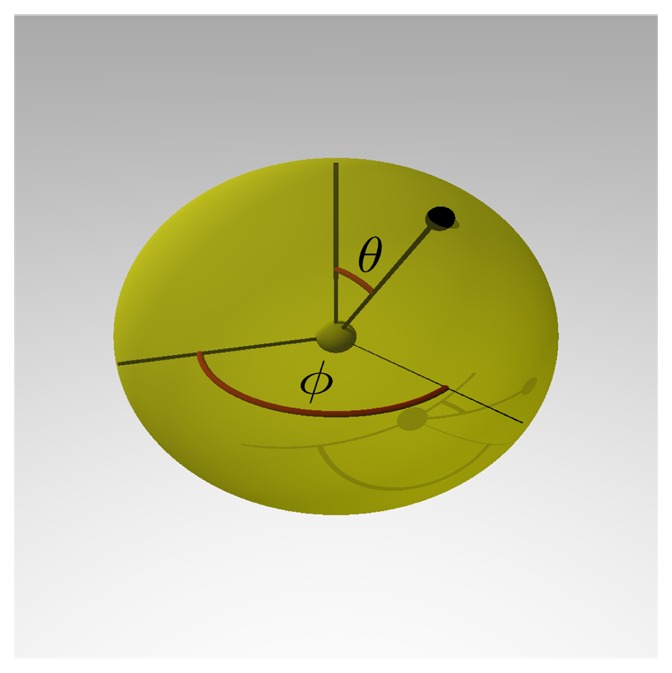
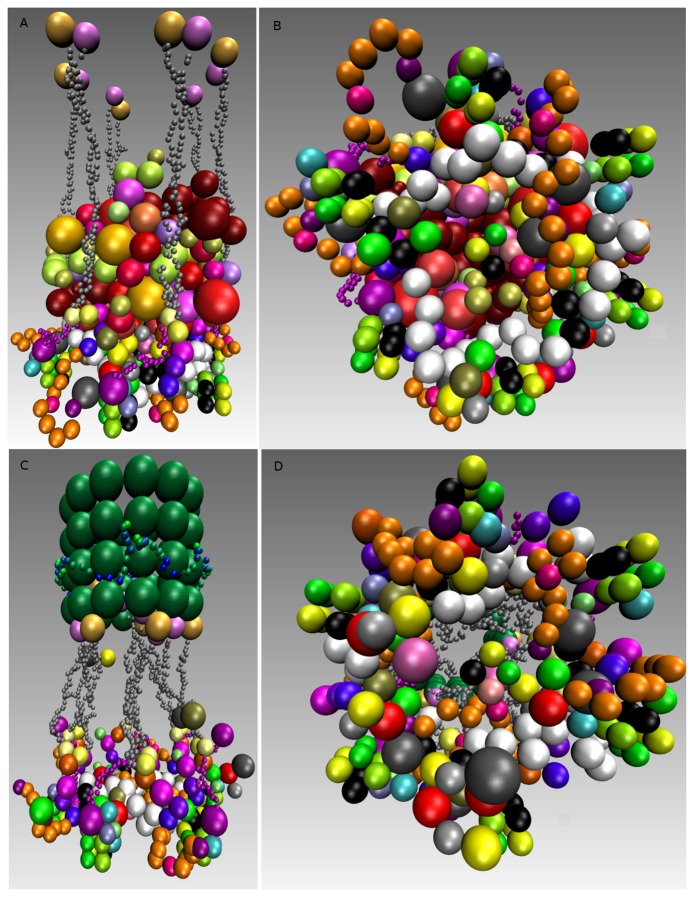
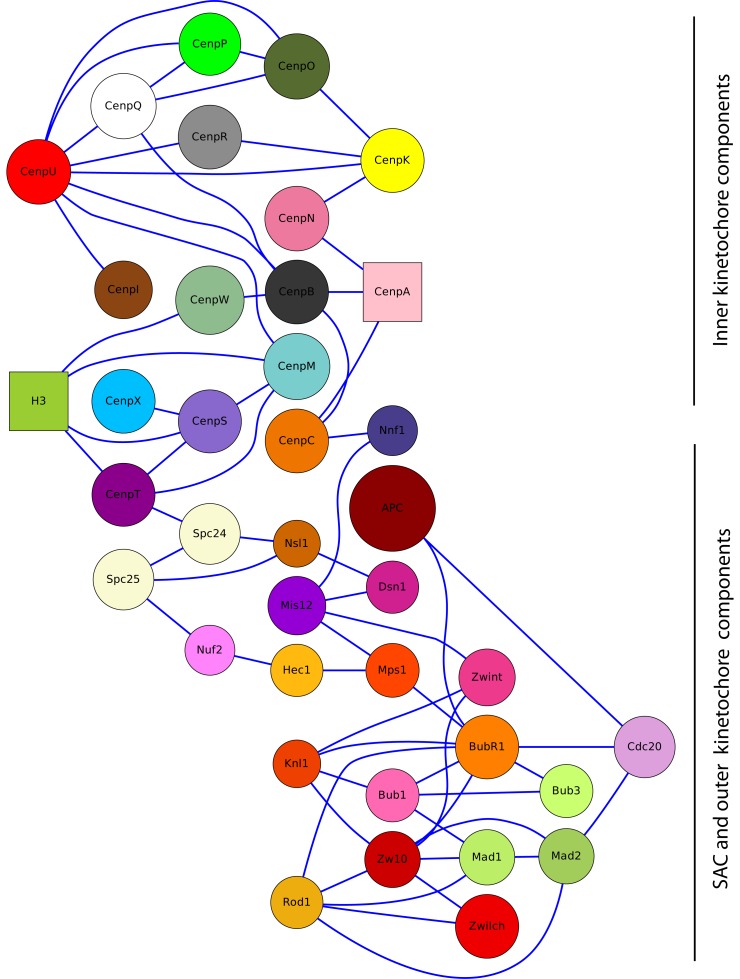
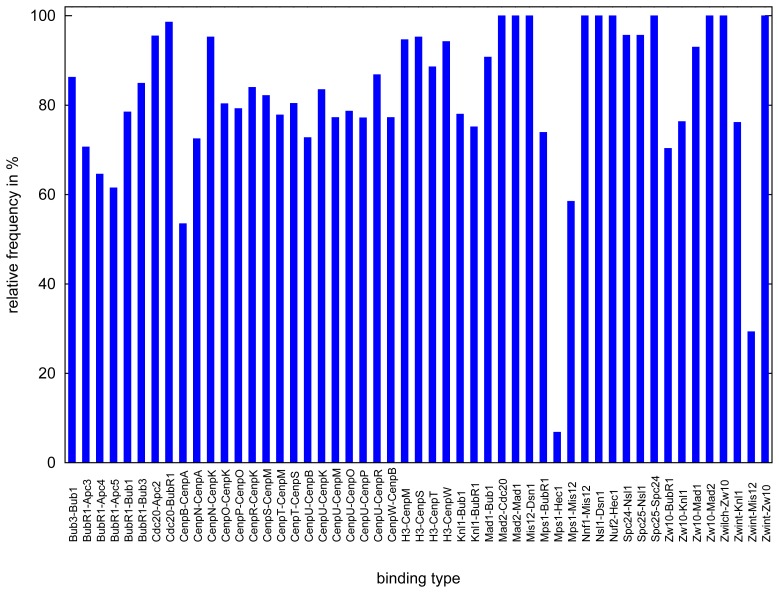
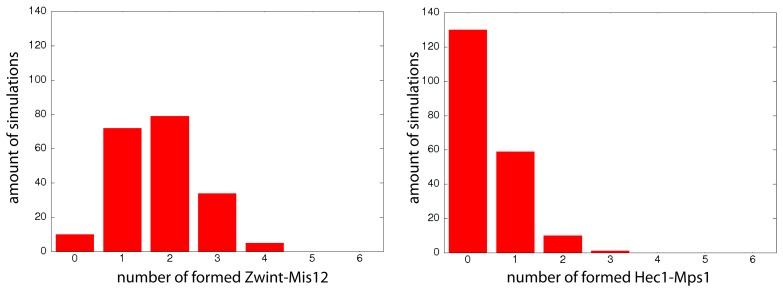
A common problem in the analysis of biological systems is the combinatorial explosion that emerges from the complexity of multi-protein assemblies. Conventional formalisms, like differential equations, Boolean networks and Bayesian networks, are unsuitable for dealing with the combinatorial explosion, because they are designed for a restricted state space with fixed dimensionality. To overcome this problem, the rule-based modeling language, BioNetGen, and the spatial extension, SRSim, have been developed. Here, we describe how to apply rule-based modeling to integrate experimental data from different sources into a single spatial simulation model and how to analyze the output of that model. The starting point for this approach can be a combination of molecular interaction data, reaction network data, proximities, binding and diffusion kinetics and molecular geometries at different levels of detail. We describe the technique and then use it to construct a model of the human mitotic inner and outer kinetochore, including the spindle assembly checkpoint signaling pathway. This allows us to demonstrate the utility of the procedure, show how a novel perspective for understanding such complex systems becomes accessible and elaborate on challenges that arise in the formulation, simulation and analysis of spatial rule-based models.
在分析生物系统时,一个常见的问题是多蛋白复合物的复杂性所导致的组合爆炸。传统的形式化方法,如微分方程、布尔网络和贝叶斯网络,不适合处理组合爆炸问题,因为它们是为具有固定维度的受限状态空间设计的。为了克服这个问题,基于规则的建模语言 BioNetGen 和空间扩展 SRSim 已经被开发出来。在这里,我们描述了如何将来自不同来源的实验数据应用于基于规则的建模,以整合到单个空间模拟模型中,以及如何分析该模型的输出。这种方法的起点可以是分子相互作用数据、反应网络数据、接近度、结合和扩散动力学以及不同细节水平的分子几何形状的组合。我们描述了该技术,然后使用它来构建人类有丝分裂内和外动粒的模型,包括纺锤体装配检查点信号通路。这使我们能够展示该过程的实用性,展示如何获得理解此类复杂系统的新视角,并详细说明在空间基于规则的模型的制定、模拟和分析中出现的挑战。