Friedrich Schiller University Jena, Bio Systems Analysis Group, 07743 Jena, Germany.
BMC Bioinformatics. 2010 Jun 7;11:307. doi: 10.1186/1471-2105-11-307.
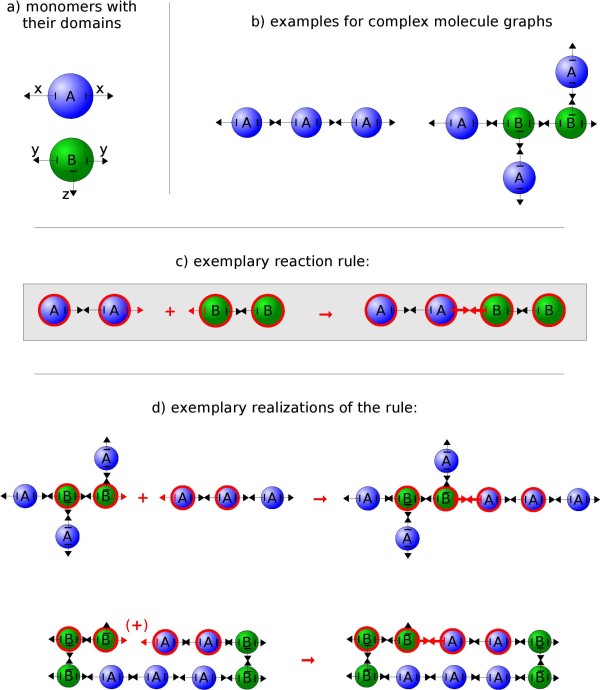
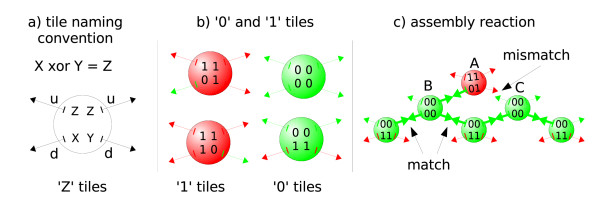
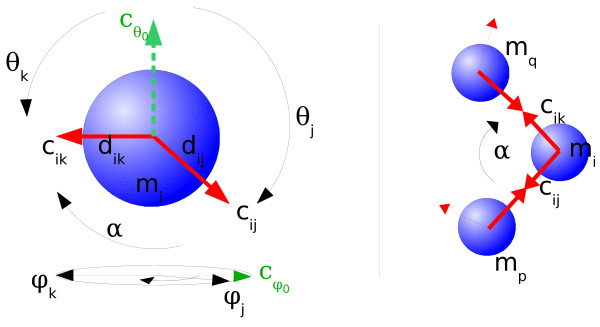
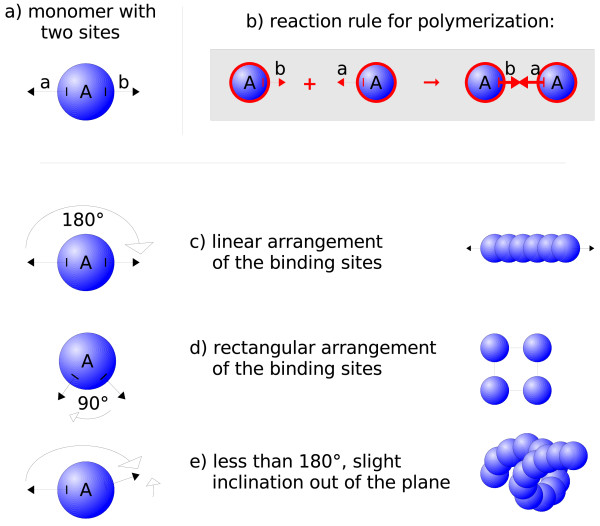
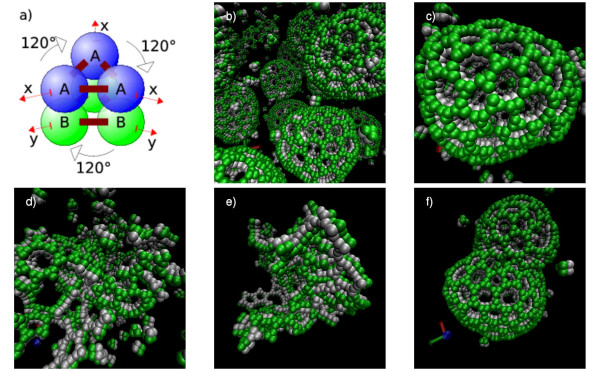
We suggest a new type of modeling approach for the coarse grained, particle-based spatial simulation of combinatorially complex chemical reaction systems. In our approach molecules possess a location in the reactor as well as an orientation and geometry, while the reactions are carried out according to a list of implicitly specified reaction rules. Because the reaction rules can contain patterns for molecules, a combinatorially complex or even infinitely sized reaction network can be defined. For our implementation (based on LAMMPS), we have chosen an already existing formalism (BioNetGen) for the implicit specification of the reaction network. This compatibility allows to import existing models easily, i.e., only additional geometry data files have to be provided.
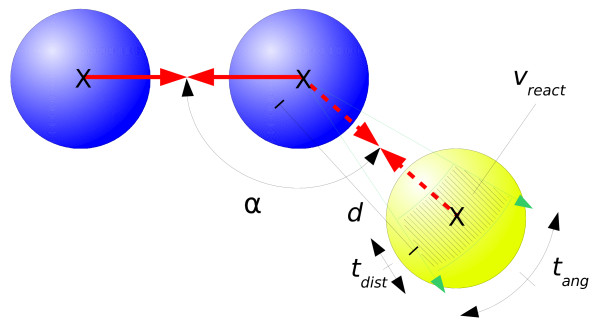
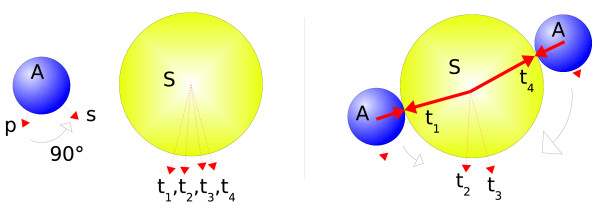
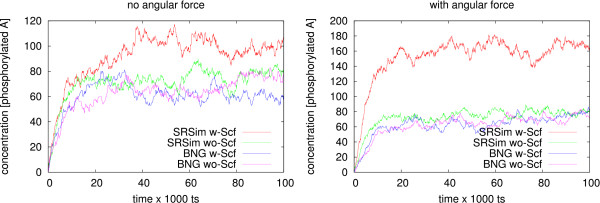
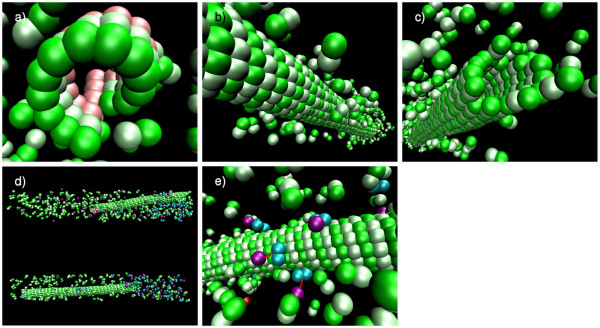
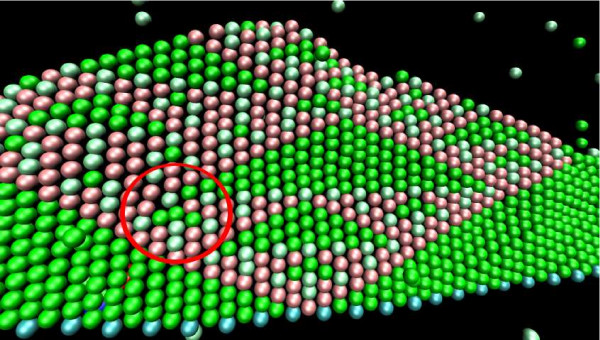
Our simulations show that the obtained dynamics can be fundamentally different from those simulations that use classical reaction-diffusion approaches like Partial Differential Equations or Gillespie-type spatial stochastic simulation. We show, for example, that the combination of combinatorial complexity and geometric effects leads to the emergence of complex self-assemblies and transportation phenomena happening faster than diffusion (using a model of molecular walkers on microtubules). When the mentioned classical simulation approaches are applied, these aspects of modeled systems cannot be observed without very special treatment. Further more, we show that the geometric information can even change the organizational structure of the reaction system. That is, a set of chemical species that can in principle form a stationary state in a Differential Equation formalism, is potentially unstable when geometry is considered, and vice versa.
We conclude that our approach provides a new general framework filling a gap in between approaches with no or rigid spatial representation like Partial Differential Equations and specialized coarse-grained spatial simulation systems like those for DNA or virus capsid self-assembly.
我们提出了一种新的建模方法,用于对组合复杂化学反应系统进行基于颗粒的粗粒度空间模拟。在我们的方法中,分子具有在反应器中的位置以及取向和几何形状,而反应根据一组隐式指定的反应规则进行。由于反应规则可以包含分子的模式,因此可以定义组合复杂甚至无限大小的反应网络。对于我们的实现(基于 LAMMPS),我们选择了一种现有的形式主义(BioNetGen)来隐式指定反应网络。这种兼容性允许轻松导入现有模型,即,仅需提供额外的几何数据文件。
我们的模拟表明,所得到的动力学可以从使用经典反应扩散方法(如偏微分方程或 Gillespie 型空间随机模拟)的模拟中根本不同。例如,我们表明,组合的复杂性和几何效应的组合导致复杂的自组装和运输现象的出现,其速度比扩散更快(使用分子在微管上行走的模型)。当应用上述经典模拟方法时,如果没有非常特殊的处理,就无法观察到模型系统的这些方面。此外,我们表明,几何信息甚至可以改变反应系统的组织结构。也就是说,在微分方程形式主义中可以原则上形成稳定状态的一组化学物质,在考虑几何形状时可能是不稳定的,反之亦然。
我们得出结论,我们的方法提供了一个新的通用框架,填补了没有或刚性空间表示的方法(如偏微分方程)和专门的粗粒度空间模拟系统(如 DNA 或病毒衣壳自组装)之间的空白。