University of Missouri-Columbia School of Medicine, Ellis Fischel Cancer Center, Columbia, MO 65212, USA.
Medical College of Georgia, Augusta, GA 30912, USA.
Genes (Basel). 2010 Jul 1;1(2):143-65. doi: 10.3390/genes1020143.
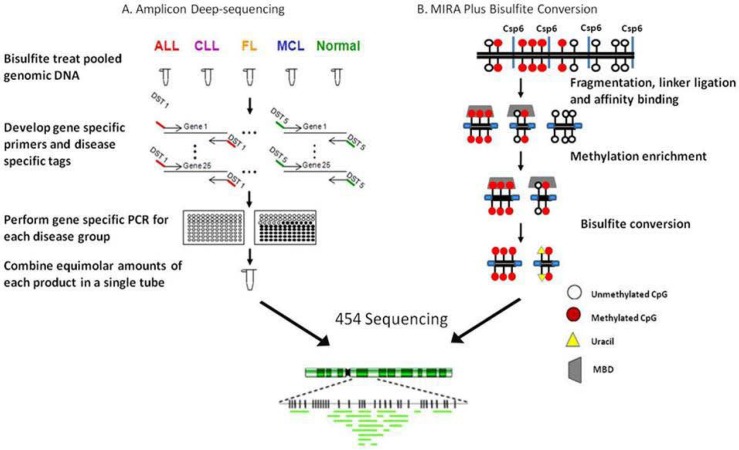
Epigenetic modifications play an important role in lymphoid malignancies. This has been evidenced by the large body of work published using microarray technologies to generate methylation profiles for numerous types and subtypes of lymphoma and leukemia. These studies have shown the importance of defining the epigenome so that we can better understand the biology of lymphoma. Recent advances in DNA sequencing technology have transformed the landscape of epigenomic analysis as we now have the ability to characterize the genome-wide distribution of chromatin modifications and DNA methylation using next-generation sequencing. To take full advantage of the throughput of next-generation sequencing, there are many methodologies that have been developed and many more that are currently being developed. Choosing the appropriate methodology is fundamental to the outcome of next-generation sequencing studies. In this review, published technologies and methodologies applicable to studying the methylome are presented. In addition, progress towards defining the methylome in lymphoma is discussed and prospective directions that have been made possible as a result of next-generation sequencing technology. Finally, methodologies are introduced that have not yet been published but that are being explored in the pursuit of defining the lymphoma methylome.
表观遗传修饰在淋巴恶性肿瘤中起着重要作用。大量的使用微阵列技术生成众多类型和亚型的淋巴瘤和白血病甲基化谱的研究已经证明了定义表观基因组的重要性,以便我们能够更好地了解淋巴瘤的生物学特性。最近 DNA 测序技术的进步改变了表观基因组分析的格局,因为我们现在有能力使用下一代测序来描述染色质修饰和 DNA 甲基化的全基因组分布。为了充分利用下一代测序的通量,已经开发了许多方法,并且还有更多的方法正在开发中。选择合适的方法对于下一代测序研究的结果至关重要。在这篇综述中,介绍了适用于研究甲基组的已发表技术和方法。此外,还讨论了在淋巴瘤中定义甲基组的进展情况,以及下一代测序技术带来的可能的新方向。最后,还介绍了一些尚未发表但正在探索中的方法,这些方法旨在定义淋巴瘤的甲基组。