Medical Systems Biology, Department of Pathology and Department of Microbiology & Immunology, University of Melbourne, Parkville, Victoria, Australia.
PLoS One. 2014 Apr 9;9(4):e93766. doi: 10.1371/journal.pone.0093766. eCollection 2014.
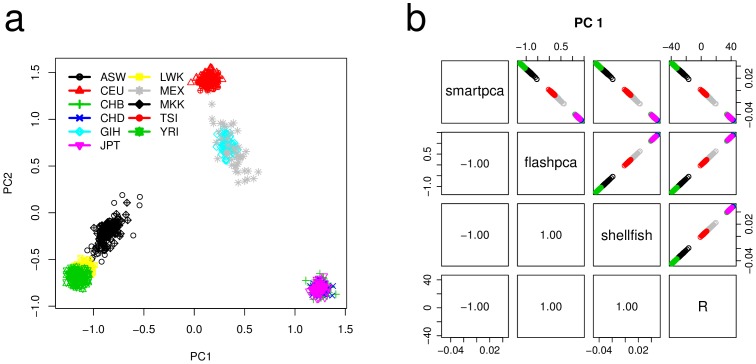
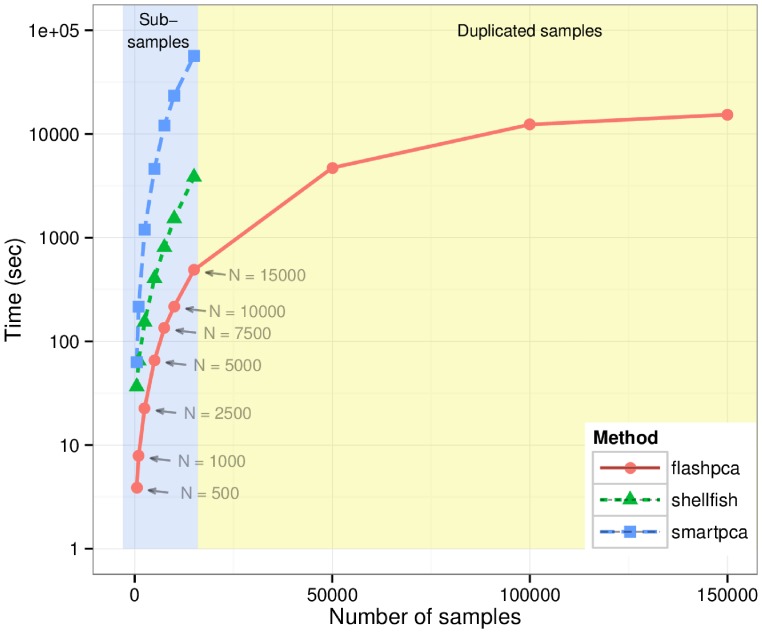
Principal component analysis (PCA) is routinely used to analyze genome-wide single-nucleotide polymorphism (SNP) data, for detecting population structure and potential outliers. However, the size of SNP datasets has increased immensely in recent years and PCA of large datasets has become a time consuming task. We have developed flashpca, a highly efficient PCA implementation based on randomized algorithms, which delivers identical accuracy in extracting the top principal components compared with existing tools, in substantially less time. We demonstrate the utility of flashpca on both HapMap3 and on a large Immunochip dataset. For the latter, flashpca performed PCA of 15,000 individuals up to 125 times faster than existing tools, with identical results, and PCA of 150,000 individuals using flashpca completed in 4 hours. The increasing size of SNP datasets will make tools such as flashpca essential as traditional approaches will not adequately scale. This approach will also help to scale other applications that leverage PCA or eigen-decomposition to substantially larger datasets.
主成分分析(PCA)通常用于分析全基因组单核苷酸多态性(SNP)数据,以检测群体结构和潜在的异常值。然而,近年来 SNP 数据集的规模已经大大增加,对大型数据集进行 PCA 已经成为一项耗时的任务。我们开发了 flashpca,这是一种基于随机算法的高效 PCA 实现方法,与现有工具相比,在提取主要成分方面具有相同的准确性,但时间大大缩短。我们在 HapMap3 和大型 Immunochip 数据集上展示了 flashpca 的实用性。对于后者,flashpca 对 15000 个人进行 PCA 的速度比现有工具快 125 倍,结果相同,而使用 flashpca 对 150000 个人进行 PCA 则在 4 小时内完成。随着 SNP 数据集规模的不断增加,像 flashpca 这样的工具将变得至关重要,因为传统方法将无法充分扩展。这种方法还将有助于将 PCA 或特征分解等其他应用扩展到更大的数据集。