From the Division of Cardiovascular and Diabetes Research, Leeds Institute of Genetics, Health and Therapeutics, Faculty of Medicine and Health and.
Institute of Membrane and Systems Biology, Faculty of Biological Sciences, University of Leeds, Leeds LS2 9JT, United Kingdom.
J Biol Chem. 2014 Jun 6;289(23):16421-9. doi: 10.1074/jbc.M114.569996. Epub 2014 Apr 9.
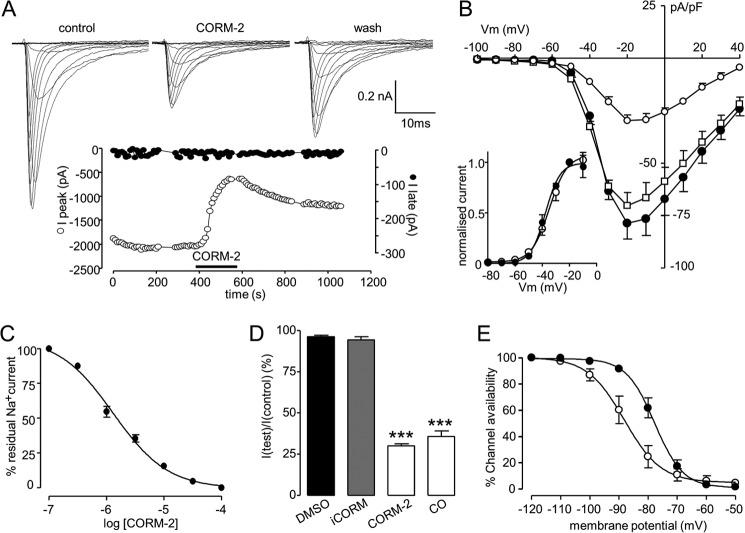
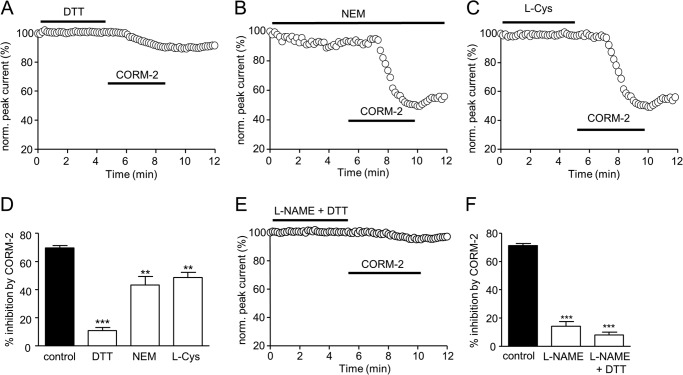
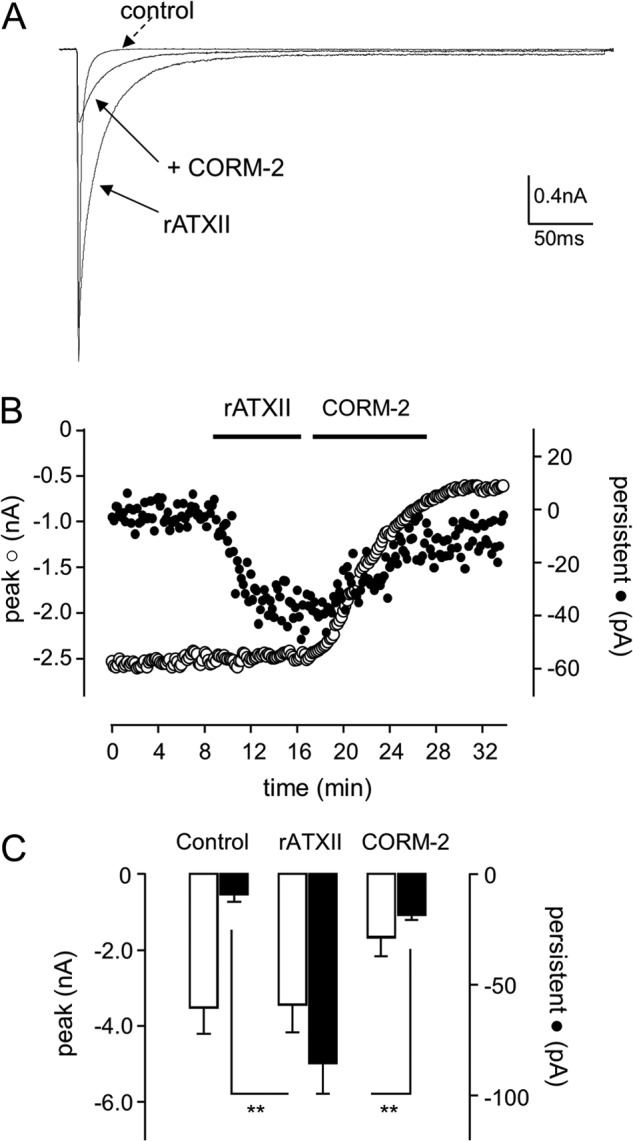
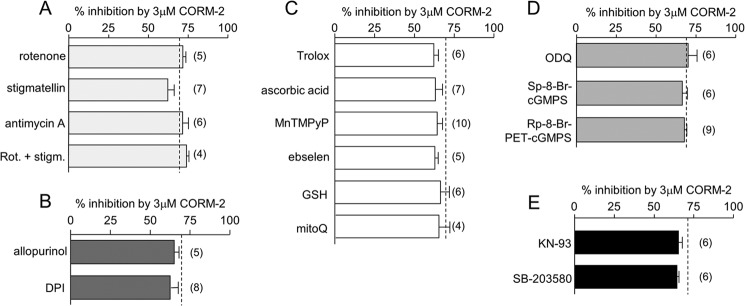
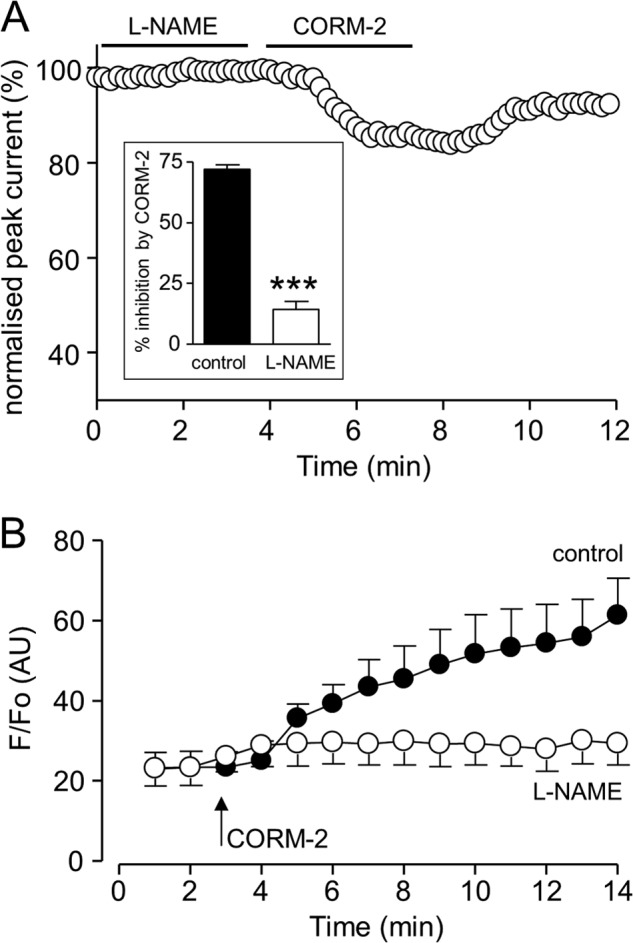
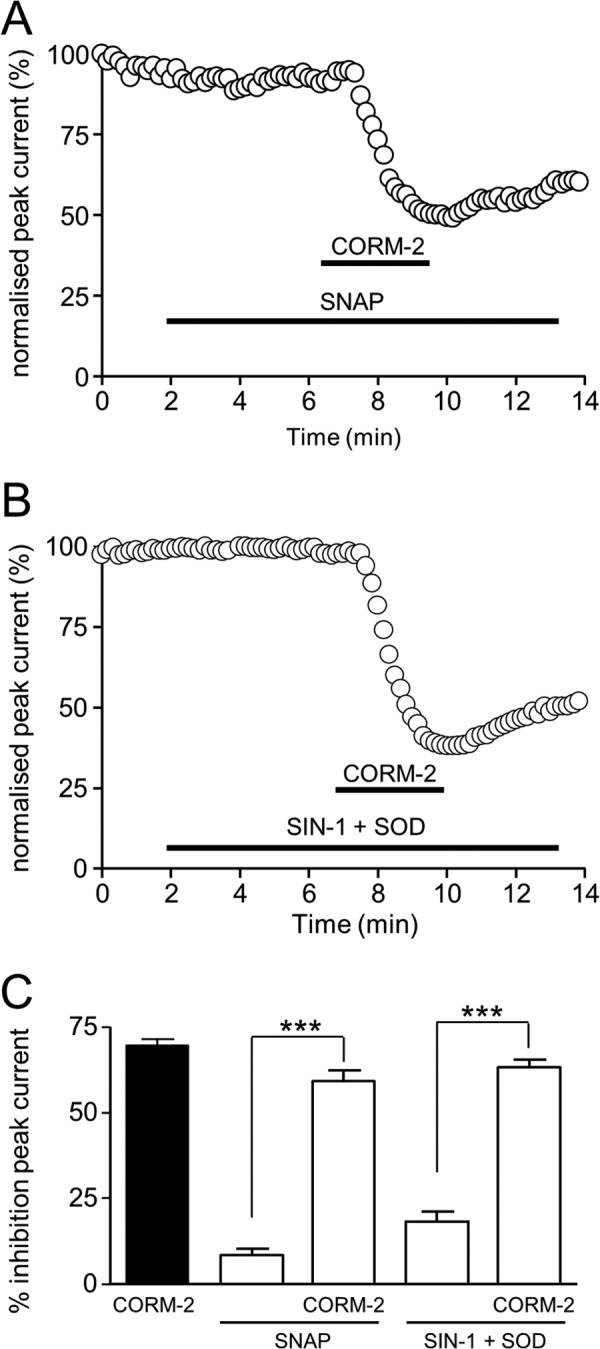
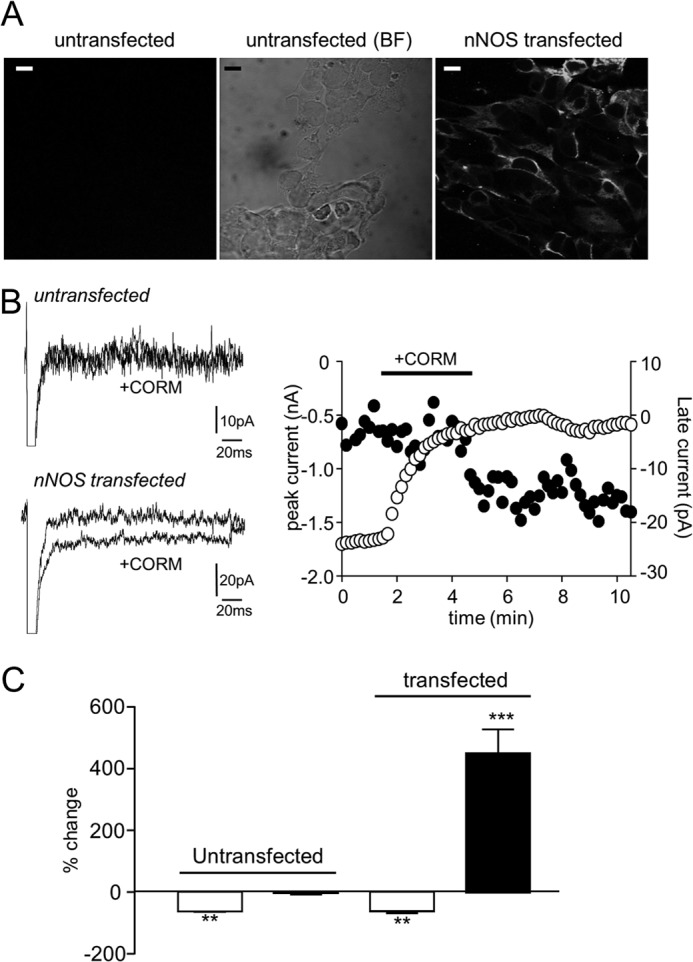
Sublethal carbon monoxide (CO) exposure is frequently associated with myocardial arrhythmias, and our recent studies have demonstrated that these may be attributable to modulation of cardiac Na(+) channels, causing an increase in the late current and an inhibition of the peak current. Using a recombinant expression system, we demonstrate that CO inhibits peak human Nav1.5 current amplitude without activation of the late Na(+) current observed in native tissue. Inhibition was associated with a hyperpolarizing shift in the steady-state inactivation properties of the channels and was unaffected by modification of channel gating induced by anemone toxin (rATX-II). Systematic pharmacological assessment indicated that no recognized CO-sensitive intracellular signaling pathways appeared to mediate CO inhibition of Nav1.5. Inhibition was, however, markedly suppressed by inhibition of NO formation, but NO donors did not mimic or occlude channel inhibition by CO, indicating that NO alone did not account for the actions of CO. Exposure of cells to DTT immediately before CO exposure also dramatically reduced the magnitude of current inhibition. Similarly, l-cysteine and N-ethylmaleimide significantly attenuated the inhibition caused by CO. In the presence of DTT and the NO inhibitor N(ω)-nitro-L-arginine methyl ester hydrochloride, the ability of CO to inhibit Nav1.5 was almost fully prevented. Our data indicate that inhibition of peak Na(+) current (which can lead to Brugada syndrome-like arrhythmias) occurs via a mechanism distinct from induction of the late current, requires NO formation, and is dependent on channel redox state.
亚致死浓度一氧化碳(CO)暴露常与心肌心律失常有关,我们最近的研究表明,这可能归因于心脏 Na+通道的调制,导致晚期电流增加和峰值电流抑制。使用重组表达系统,我们证明 CO 抑制了人 Nav1.5 峰值电流幅度,而没有激活天然组织中观察到的晚期 Na+电流。抑制与通道稳态失活特性的超极化偏移有关,不受海葵毒素(rATX-II)诱导的通道门控修饰的影响。系统药理学评估表明,没有发现与 CO 抑制 Nav1.5 相关的公认的 CO 敏感细胞内信号通路。然而,NO 形成的抑制显著抑制了抑制作用,但 NO 供体不能模拟或阻断 CO 对通道的抑制,表明 NO 本身不能解释 CO 的作用。在 CO 暴露之前立即将细胞暴露于 DTT 也大大降低了电流抑制的幅度。同样,l-半胱氨酸和 N-乙基马来酰亚胺显著减弱了 CO 引起的抑制作用。在 DTT 和 NO 抑制剂 N(ω)-硝基-L-精氨酸甲酯盐酸盐存在的情况下,CO 抑制 Nav1.5 的能力几乎完全被阻止。我们的数据表明,峰值 Na+电流的抑制(可导致 Brugada 综合征样心律失常)通过不同于诱导晚期电流的机制发生,需要 NO 形成,并取决于通道氧化还原状态。