de Jong Johann, Akhtar Waseem, Badhai Jitendra, Rust Alistair G, Rad Roland, Hilkens John, Berns Anton, van Lohuizen Maarten, Wessels Lodewyk F A, de Ridder Jeroen
Computational Cancer Biology Group, Division of Molecular Carcinogenesis, The Netherlands Cancer Institute, Amsterdam, The Netherlands; Netherlands Consortium for Systems Biology, Amsterdam, The Netherlands.
Netherlands Consortium for Systems Biology, Amsterdam, The Netherlands; Division of Molecular Genetics, The Netherlands Cancer Institute, Amsterdam, The Netherlands.
PLoS Genet. 2014 Apr 10;10(4):e1004250. doi: 10.1371/journal.pgen.1004250. eCollection 2014 Apr.
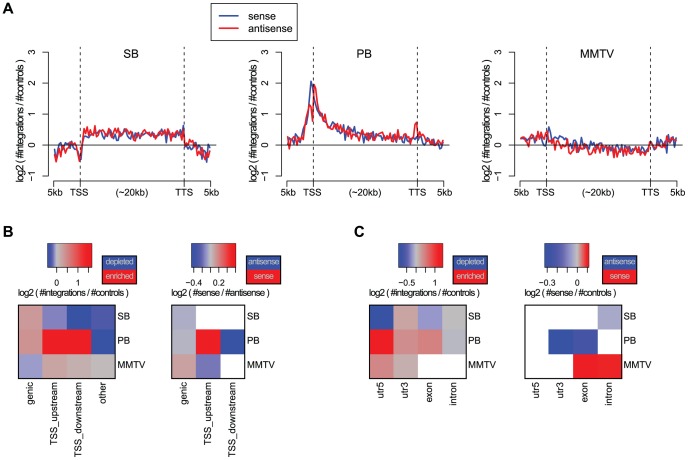
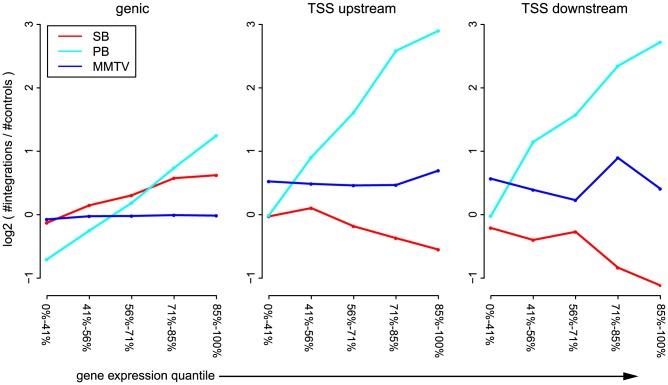
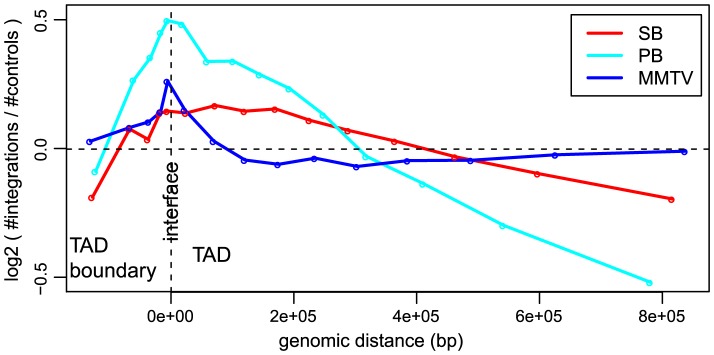
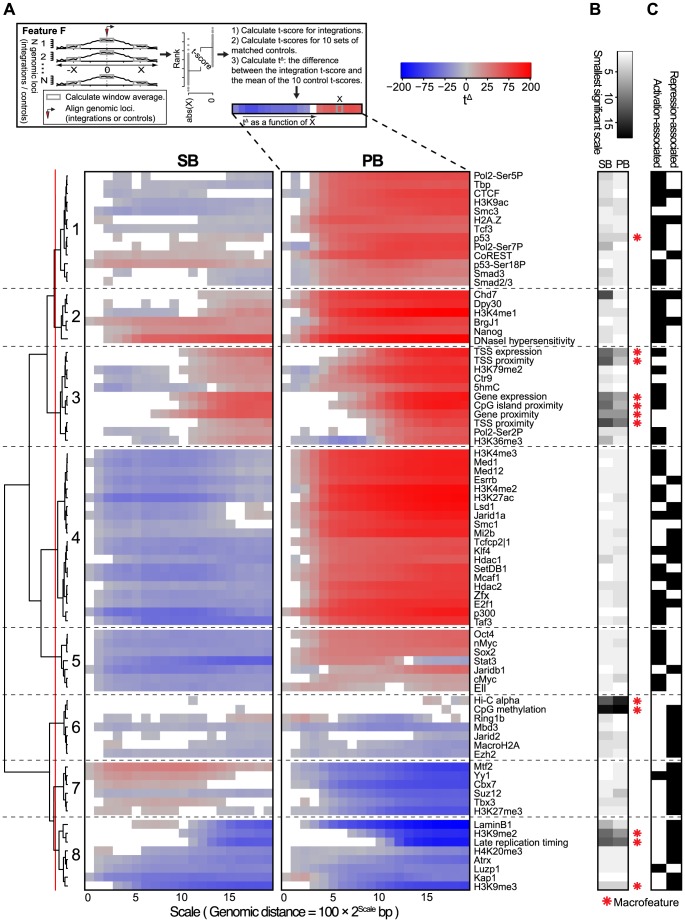
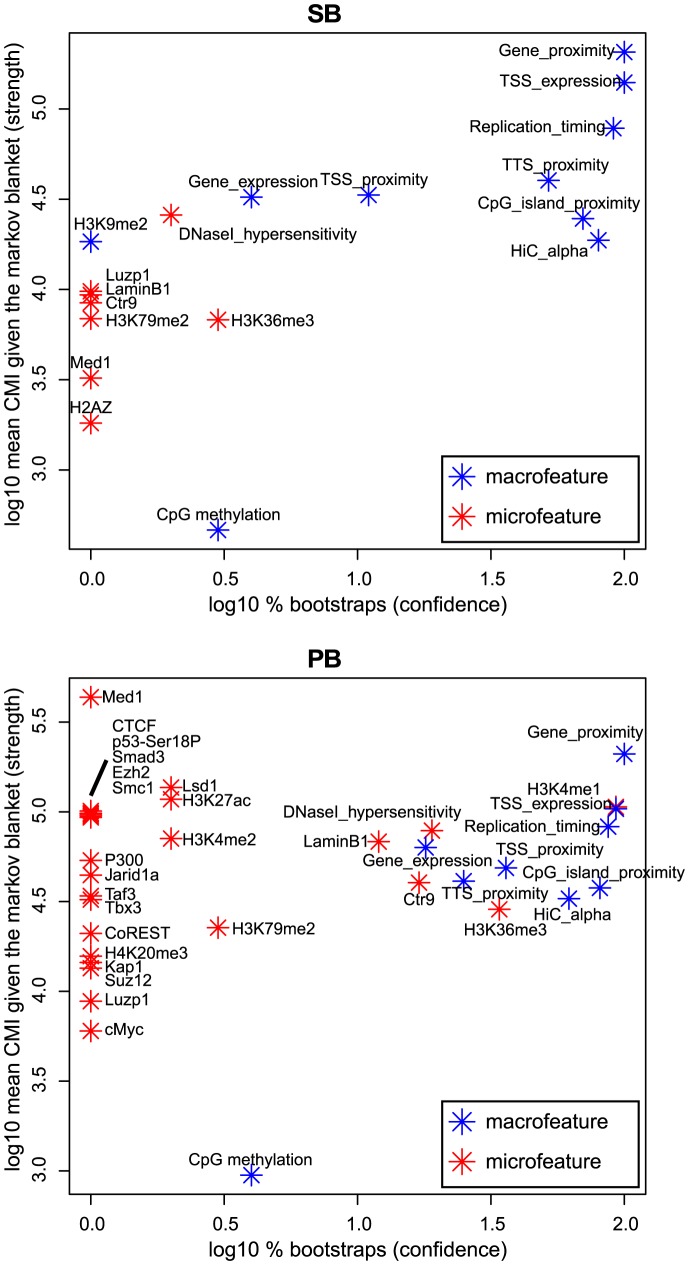
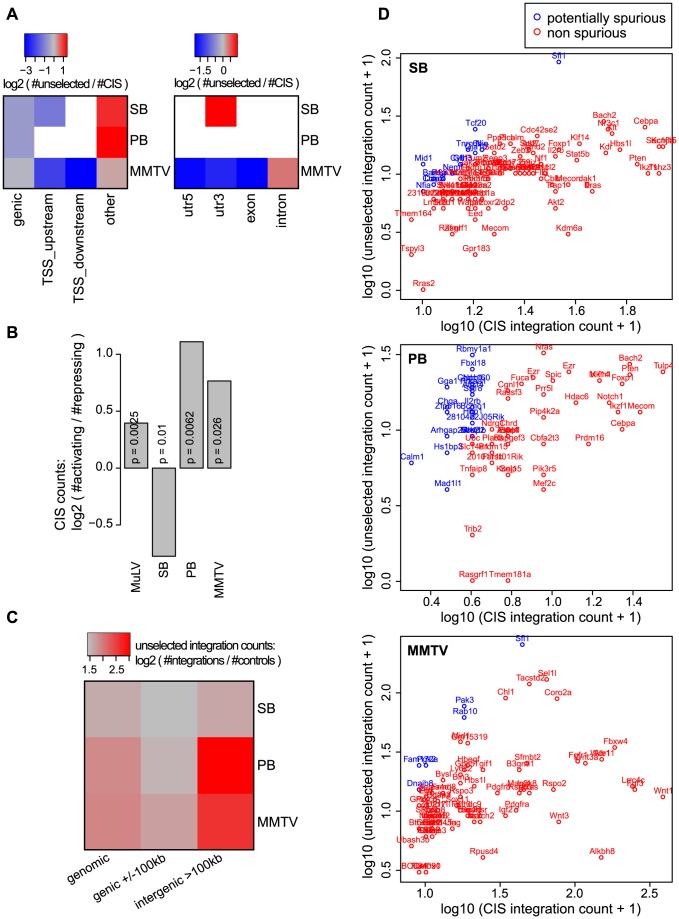
The ability of retroviruses and transposons to insert their genetic material into host DNA makes them widely used tools in molecular biology, cancer research and gene therapy. However, these systems have biases that may strongly affect research outcomes. To address this issue, we generated very large datasets consisting of ~ 120,000 to ~ 180,000 unselected integrations in the mouse genome for the Sleeping Beauty (SB) and piggyBac (PB) transposons, and the Mouse Mammary Tumor Virus (MMTV). We analyzed ~ 80 (epi)genomic features to generate bias maps at both local and genome-wide scales. MMTV showed a remarkably uniform distribution of integrations across the genome. More distinct preferences were observed for the two transposons, with PB showing remarkable resemblance to bias profiles of the Murine Leukemia Virus. Furthermore, we present a model where target site selection is directed at multiple scales. At a large scale, target site selection is similar across systems, and defined by domain-oriented features, namely expression of proximal genes, proximity to CpG islands and to genic features, chromatin compaction and replication timing. Notable differences between the systems are mainly observed at smaller scales, and are directed by a diverse range of features. To study the effect of these biases on integration sites occupied under selective pressure, we turned to insertional mutagenesis (IM) screens. In IM screens, putative cancer genes are identified by finding frequently targeted genomic regions, or Common Integration Sites (CISs). Within three recently completed IM screens, we identified 7%-33% putative false positive CISs, which are likely not the result of the oncogenic selection process. Moreover, results indicate that PB, compared to SB, is more suited to tag oncogenes.
逆转录病毒和转座子能够将其遗传物质插入宿主DNA,这使得它们成为分子生物学、癌症研究和基因治疗中广泛使用的工具。然而,这些系统存在偏差,可能会强烈影响研究结果。为了解决这个问题,我们生成了非常大的数据集,其中包含小鼠基因组中约120,000至约180,000个未选择的睡美人(SB)转座子、猪尾巴(PB)转座子和小鼠乳腺肿瘤病毒(MMTV)的整合。我们分析了约80个(表观)基因组特征,以在局部和全基因组尺度上生成偏差图谱。MMTV在全基因组中的整合分布非常均匀。在两个转座子中观察到了更明显的偏好,PB与鼠白血病病毒的偏差图谱有显著相似之处。此外,我们提出了一个模型,其中靶位点选择是在多个尺度上进行的。在大尺度上,不同系统的靶位点选择相似,并由面向结构域的特征定义,即近端基因的表达、与CpG岛和基因特征的接近程度、染色质压缩和复制时间。不同系统之间的显著差异主要在较小尺度上观察到,并由多种特征决定。为了研究这些偏差对在选择压力下占据的整合位点的影响,我们转向插入诱变(IM)筛选。在IM筛选中,通过找到频繁靶向的基因组区域或共同整合位点(CIS)来鉴定潜在的癌症基因。在最近完成的三项IM筛选中,我们鉴定出7%-33%的潜在假阳性CIS,它们可能不是致癌选择过程的结果。此外,结果表明,与SB相比,PB更适合标记致癌基因。