Faculty of Computing, Engineering and Sciences, Staffordshire University, Stoke-on-Trent, United Kingdom.
Department of Infection, Immunity and Inflammation, Institute for Lung Health, University of Leicester, Leicester, United Kingdom.
PLoS One. 2014 Apr 11;9(4):e93849. doi: 10.1371/journal.pone.0093849. eCollection 2014.
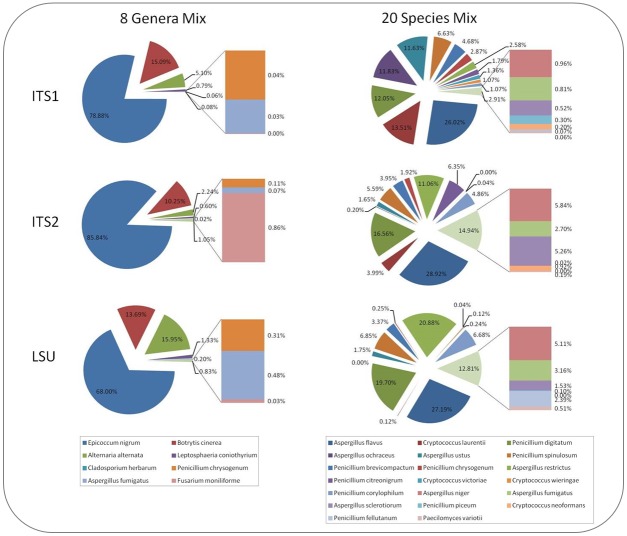
Next generation sequencing technology has revolutionised microbiology by allowing concurrent analysis of whole microbial communities. Here we developed and verified similar methods for the analysis of fungal communities using a proton release sequencing platform with the ability to sequence reads of up to 400 bp in length at significant depth. This read length permits the sequencing of amplicons from commonly used fungal identification regions and thereby taxonomic classification. Using the 400 bp sequencing capability, we have sequenced amplicons from the ITS1, ITS2 and LSU fungal regions to a depth of approximately 700,000 raw reads per sample. Representative operational taxonomic units (OTUs) were chosen by the USEARCH algorithm, and identified taxonomically through nucleotide blast (BLASTn). Combination of this sequencing technology with the bioinformatics pipeline allowed species recognition in two controlled fungal spore populations containing members of known identity and concentration. Each species included within the two controlled populations was found to correspond to a representative OTU, and these OTUs were found to be highly accurate representations of true biological sequences. However, the absolute number of reads attributed to each OTU differed among species. The majority of species were represented by an OTU derived from all three genomic regions although in some cases, species were only represented in two of the regions due to the absence of conserved primer binding sites or due to sequence composition. It is apparent from our data that proton release sequencing technologies can deliver a qualitative assessment of the fungal members comprising a sample. The fact that some fungi cannot be amplified by specific "conserved" primer pairs confirms our recommendation that a multi-region approach be taken for other amplicon-based metagenomic studies.
下一代测序技术通过同时分析整个微生物群落,彻底改变了微生物学。在这里,我们开发并验证了类似的方法,用于使用具有能够以显著深度测序长达 400 个碱基对的读长的质子释放测序平台分析真菌群落。该读长允许对常用真菌鉴定区域的扩增子进行测序,从而进行分类学分类。使用 400 个碱基对测序能力,我们已经对 ITS1、ITS2 和 LSU 真菌区域的扩增子进行了测序,每个样本的原始读数深度约为 700,000。代表性操作分类单元 (OTU) 通过 USEARCH 算法选择,并通过核苷酸比对 (BLASTn) 进行分类鉴定。将这种测序技术与生物信息学管道相结合,允许在包含已知身份和浓度成员的两个受控真菌孢子群体中识别物种。包含在两个受控群体中的每个物种都被发现对应于一个代表 OTU,并且这些 OTU 被发现是真实生物序列的高度准确表示。然而,每个 OTU 归因于的读数值在物种之间存在差异。大多数物种都由源自所有三个基因组区域的 OTU 表示,尽管在某些情况下,由于缺乏保守的引物结合位点或由于序列组成,某些物种仅在两个区域中被代表。从我们的数据中可以明显看出,质子释放测序技术可以对构成样本的真菌成员进行定性评估。有些真菌不能被特定的“保守”引物对扩增的事实证实了我们的建议,即对于其他基于扩增子的宏基因组研究,应采用多区域方法。