Beier Rico, Boschke Elke, Labudde Dirk
Bioinformatics Group, Department of Mathematics, Natural and Computer Sciences, University of Applied Sciences Mittweida, 09648 Mittweida, Germany.
Institute of Food Technology and Bioprocess Engineering, Department of Mechanical Engineering, Dresden University of Technology, 01062 Dresden, Germany.
Biomed Res Int. 2014;2014:849743. doi: 10.1155/2014/849743. Epub 2014 Mar 19.
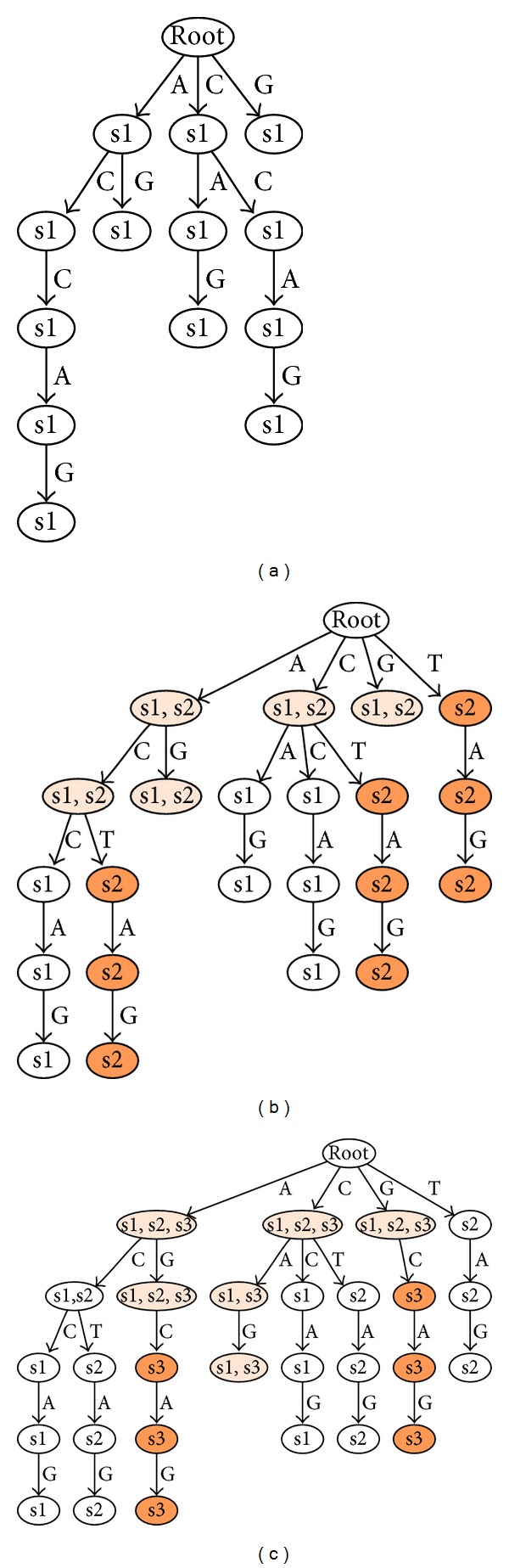
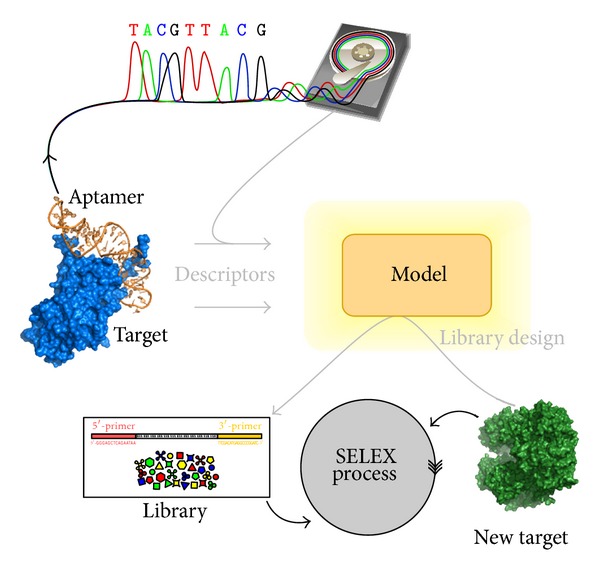
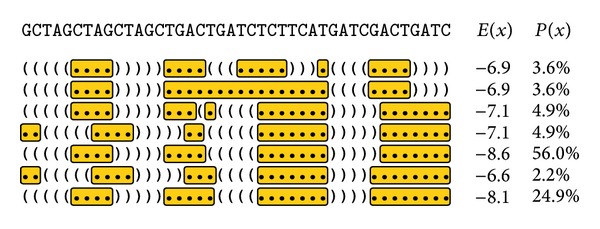
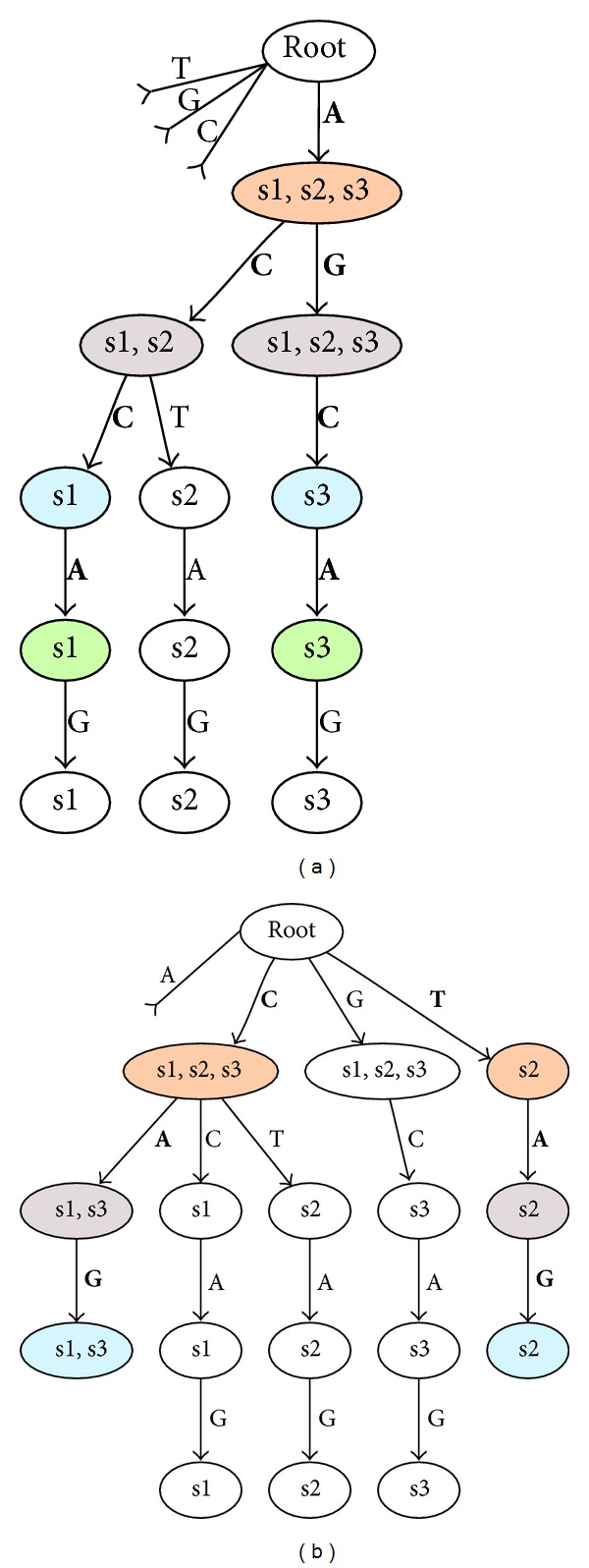
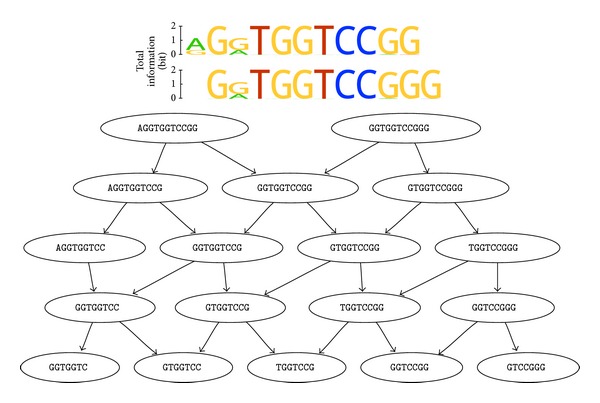
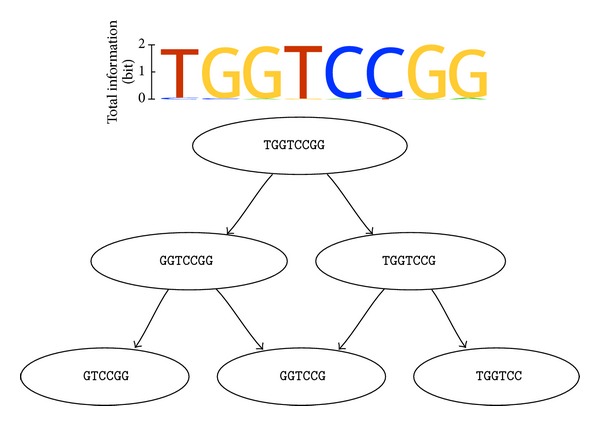
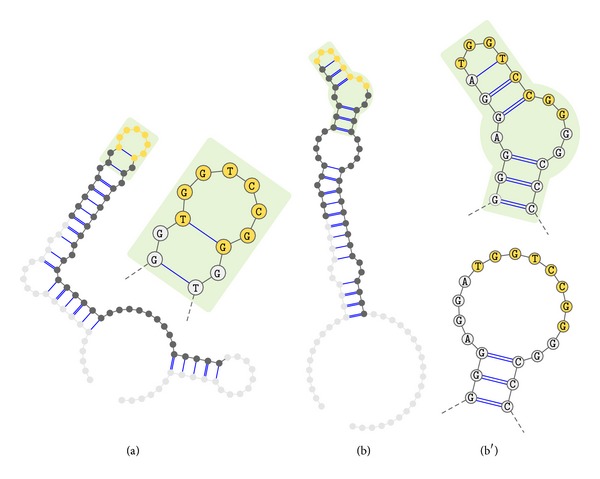
Aptamers are an interesting alternative to antibodies in pharmaceutics and biosensorics, because they are able to bind to a multitude of possible target molecules with high affinity. Therefore the process of finding such aptamers, which is commonly a SELEX screening process, becomes crucial. The standard SELEX procedure schedules the validation of certain found aptamers via binding experiments, which is not leading to any detailed specification of the aptamer enrichment during the screening. For the purpose of advanced analysis of the accrued enrichment within the SELEX library we used sequence information gathered by next generation sequencing techniques in addition to the standard SELEX procedure. As sequence motifs are one possibility of enrichment description, the need of finding those recurring sequence motifs corresponding to substructures within the aptamers, which are characteristically fitted to specific binding sites of the target, arises. In this paper a motif search algorithm is presented, which helps to describe the aptamers enrichment in more detail. The extensive characterization of target and binding aptamers may later reveal a functional connection between these molecules, which can be modeled and used to optimize future SELEX runs in case of the generation of target-specific starting libraries.
适体在制药学和生物传感领域是一种有趣的抗体替代物,因为它们能够以高亲和力结合多种可能的靶分子。因此,寻找此类适体的过程(通常是指数富集的配体系统进化筛选过程)变得至关重要。标准的指数富集的配体系统进化程序通过结合实验对某些发现的适体进行验证,但这在筛选过程中并不能对适体富集进行任何详细的说明。为了对指数富集的配体系统进化文库中累积的富集进行深入分析,除了标准的指数富集的配体系统进化程序外,我们还使用了通过下一代测序技术收集的序列信息。由于序列基序是富集描述的一种方式,因此需要找到那些与适体内的子结构相对应的重复序列基序,这些子结构特征性地适合于靶标的特定结合位点。本文提出了一种基序搜索算法,有助于更详细地描述适体的富集情况。对靶标和结合适体的广泛表征可能随后揭示这些分子之间的功能联系,在生成靶标特异性起始文库时,这种联系可以被建模并用于优化未来的指数富集的配体系统进化实验。