Adoue Veronique, Schiavi Alicia, Light Nicholas, Almlöf Jonas Carlsson, Lundmark Per, Ge Bing, Kwan Tony, Caron Maxime, Rönnblom Lars, Wang Chuan, Chen Shu-Huang, Goodall Alison H, Cambien Francois, Deloukas Panos, Ouwehand Willem H, Syvänen Ann-Christine, Pastinen Tomi
Institute National de la Santé et de la Recherche Médicale (INSERM), U1043, Toulouse, France.
Department of Human Genetics, McGill University and Genome Quebec Innovation Centre, Montreal, QC, Canada.
Mol Syst Biol. 2014 Oct 16;10(10):754. doi: 10.15252/msb.20145114.
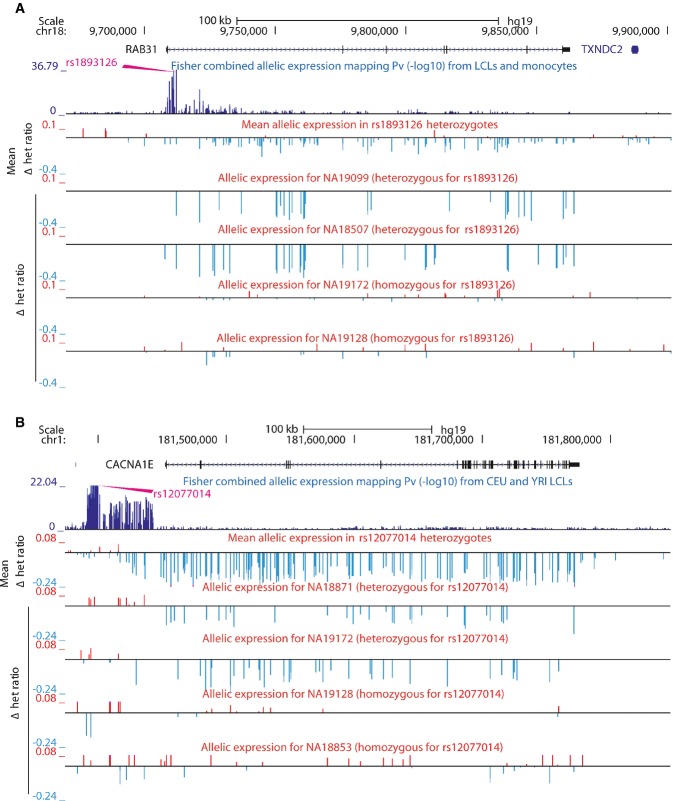
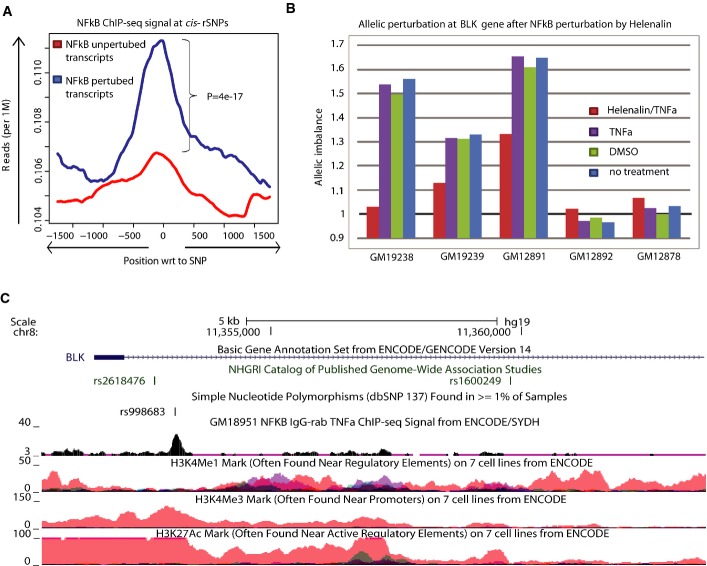
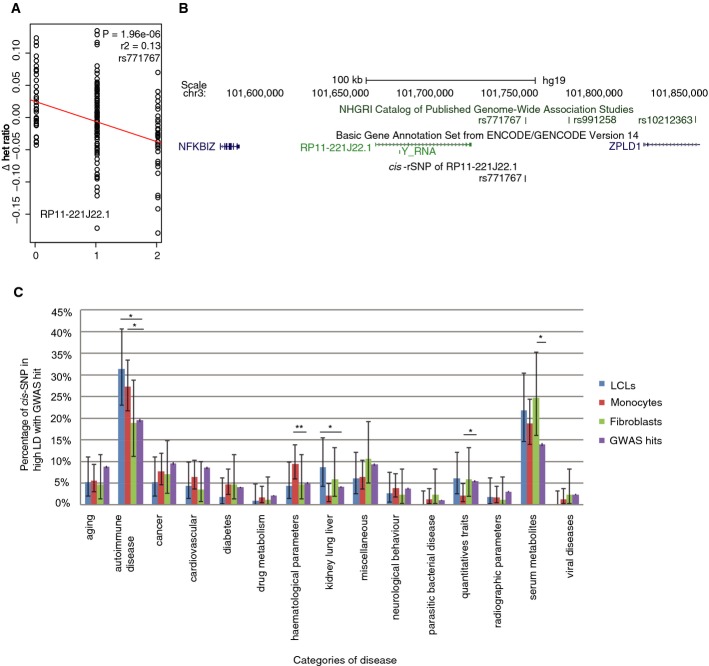
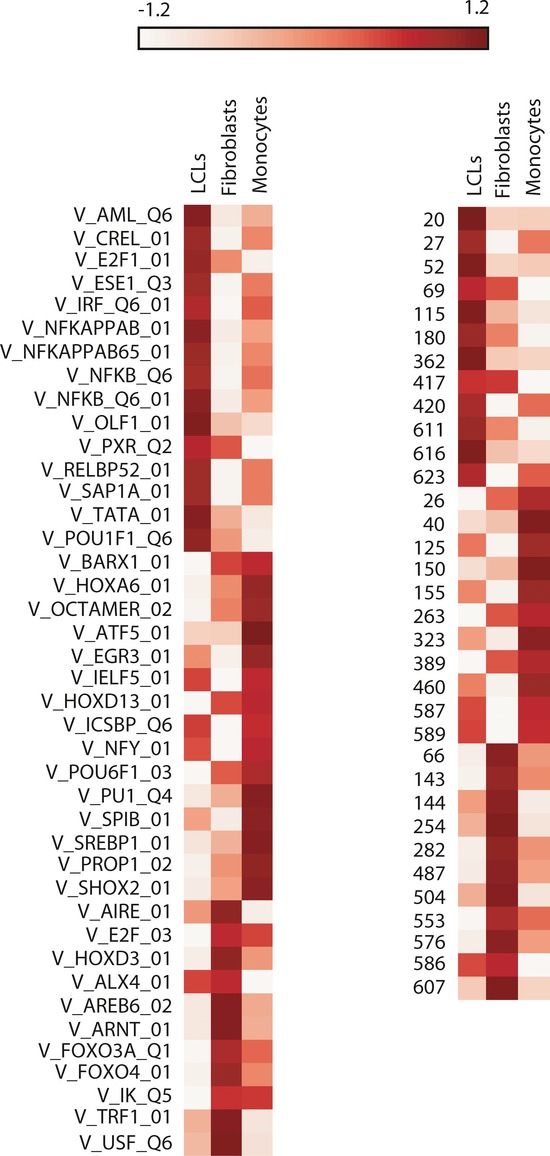
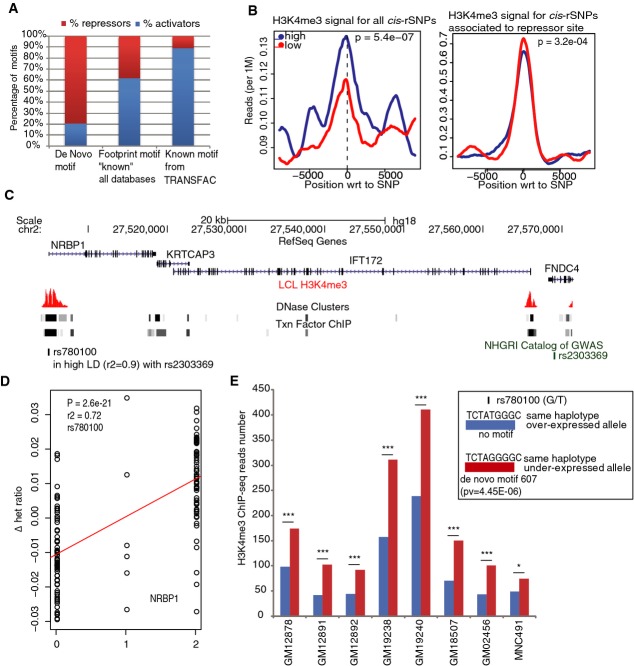
Most complex disease-associated genetic variants are located in non-coding regions and are therefore thought to be regulatory in nature. Association mapping of differential allelic expression (AE) is a powerful method to identify SNPs with direct cis-regulatory impact (cis-rSNPs). We used AE mapping to identify cis-rSNPs regulating gene expression in 55 and 63 HapMap lymphoblastoid cell lines from a Caucasian and an African population, respectively, 70 fibroblast cell lines, and 188 purified monocyte samples and found 40-60% of these cis-rSNPs to be shared across cell types. We uncover a new class of cis-rSNPs, which disrupt footprint-derived de novo motifs that are predominantly bound by repressive factors and are implicated in disease susceptibility through overlaps with GWAS SNPs. Finally, we provide the proof-of-principle for a new approach for genome-wide functional validation of transcription factor-SNP interactions. By perturbing NFκB action in lymphoblasts, we identified 489 cis-regulated transcripts with altered AE after NFκB perturbation. Altogether, we perform a comprehensive analysis of cis-variation in four cell populations and provide new tools for the identification of functional variants associated to complex diseases.
大多数与复杂疾病相关的基因变异位于非编码区域,因此被认为本质上具有调控作用。差异等位基因表达(AE)的关联图谱分析是一种强大的方法,用于识别具有直接顺式调控影响的单核苷酸多态性(cis-rSNP)。我们分别使用AE图谱分析,在来自高加索人群和非洲人群的55个和63个HapMap淋巴母细胞系、70个成纤维细胞系以及188个纯化单核细胞样本中识别调控基因表达的cis-rSNP,发现这些cis-rSNP中有40%-60%在不同细胞类型中是共享的。我们发现了一类新的cis-rSNP,它们破坏了源自足迹的新生基序,这些基序主要由抑制因子结合,并通过与全基因组关联研究(GWAS)单核苷酸多态性重叠而与疾病易感性相关。最后,我们为转录因子-单核苷酸多态性相互作用的全基因组功能验证新方法提供了原理证明。通过干扰淋巴母细胞中的核因子κB(NFκB)作用,我们在NFκB干扰后鉴定出489个AE发生改变的顺式调控转录本。总之,我们对四个细胞群体中的顺式变异进行了全面分析,并为识别与复杂疾病相关的功能变异提供了新工具。