Ma Shuyi, Sung Jaeyun, Magis Andrew T, Wang Yuliang, Geman Donald, Price Nathan D
Institute for Systems Biology, Seattle, Washington, United States of America; Department of Chemical and Biomolecular Engineering, University of Illinois, Urbana, Illinois, United States of America.
Institute for Systems Biology, Seattle, Washington, United States of America; Asia Pacific Center for Theoretical Physics, Pohang, Gyeongbuk, Republic of Korea.
PLoS One. 2014 Oct 17;9(10):e110840. doi: 10.1371/journal.pone.0110840. eCollection 2014.
The biomarker discovery field is replete with molecular signatures that have not translated into the clinic despite ostensibly promising performance in predicting disease phenotypes. One widely cited reason is lack of classification consistency, largely due to failure to maintain performance from study to study. This failure is widely attributed to variability in data collected for the same phenotype among disparate studies, due to technical factors unrelated to phenotypes (e.g., laboratory settings resulting in "batch-effects") and non-phenotype-associated biological variation in the underlying populations. These sources of variability persist in new data collection technologies.
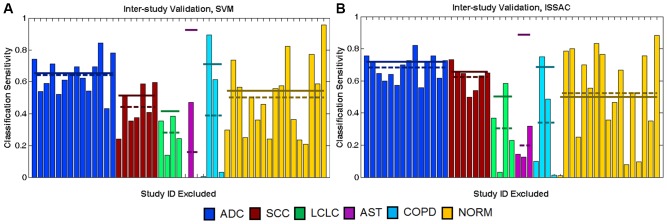
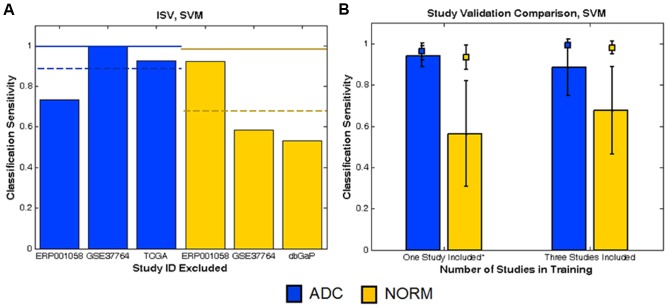
Here we quantify the impact of these combined "study-effects" on a disease signature's predictive performance by comparing two types of validation methods: ordinary randomized cross-validation (RCV), which extracts random subsets of samples for testing, and inter-study validation (ISV), which excludes an entire study for testing. Whereas RCV hardwires an assumption of training and testing on identically distributed data, this key property is lost in ISV, yielding systematic decreases in performance estimates relative to RCV. Measuring the RCV-ISV difference as a function of number of studies quantifies influence of study-effects on performance.
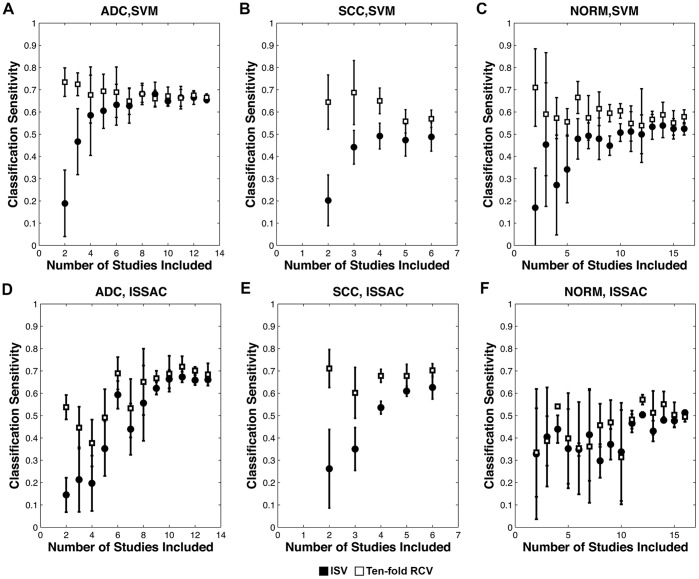
As a case study, we gathered publicly available gene expression data from 1,470 microarray samples of 6 lung phenotypes from 26 independent experimental studies and 769 RNA-seq samples of 2 lung phenotypes from 4 independent studies. We find that the RCV-ISV performance discrepancy is greater in phenotypes with few studies, and that the ISV performance converges toward RCV performance as data from additional studies are incorporated into classification.
We show that by examining how fast ISV performance approaches RCV as the number of studies is increased, one can estimate when "sufficient" diversity has been achieved for learning a molecular signature likely to translate without significant loss of accuracy to new clinical settings.
生物标志物发现领域充斥着各种分子特征,尽管在预测疾病表型方面表面上表现出有前景的性能,但尚未转化为临床应用。一个被广泛引用的原因是缺乏分类一致性,这主要是由于不同研究之间未能保持性能。这种失败被广泛归因于不同研究中针对同一表型收集的数据存在变异性,这是由与表型无关的技术因素(例如导致“批次效应”的实验室设置)以及基础人群中与表型无关的生物学变异造成的。这些变异性来源在新的数据收集技术中依然存在。
在此,我们通过比较两种验证方法来量化这些综合“研究效应”对疾病特征预测性能的影响:普通随机交叉验证(RCV),它提取随机样本子集进行测试;以及跨研究验证(ISV),它排除整个研究进行测试。虽然RCV硬性假定在同分布数据上进行训练和测试,但在ISV中这个关键属性丧失了,导致相对于RCV,性能估计出现系统性下降。将RCV - ISV差异作为研究数量的函数进行测量,可量化研究效应对性能的影响。
作为一个案例研究,我们从26项独立实验研究的1470个6种肺表型的微阵列样本以及4项独立研究的769个2种肺表型的RNA测序样本中收集了公开可用的基因表达数据。我们发现,在研究较少的表型中,RCV - ISV性能差异更大,并且随着来自更多研究的数据被纳入分类,ISV性能趋向于RCV性能。
我们表明,通过检查随着研究数量增加ISV性能接近RCV的速度,人们可以估计何时已经实现了“足够”的多样性,以便学习到一个可能在不显著损失准确性的情况下转化到新临床环境的分子特征。