Chung Neo Christopher, Storey John D
Lewis-Sigler Institute for Integrative Genomics and Department of Molecular Biology, Princeton University, Princeton, NJ 08544, USA.
Lewis-Sigler Institute for Integrative Genomics and Department of Molecular Biology, Princeton University, Princeton, NJ 08544, USA Lewis-Sigler Institute for Integrative Genomics and Department of Molecular Biology, Princeton University, Princeton, NJ 08544, USA.
Bioinformatics. 2015 Feb 15;31(4):545-54. doi: 10.1093/bioinformatics/btu674. Epub 2014 Oct 21.
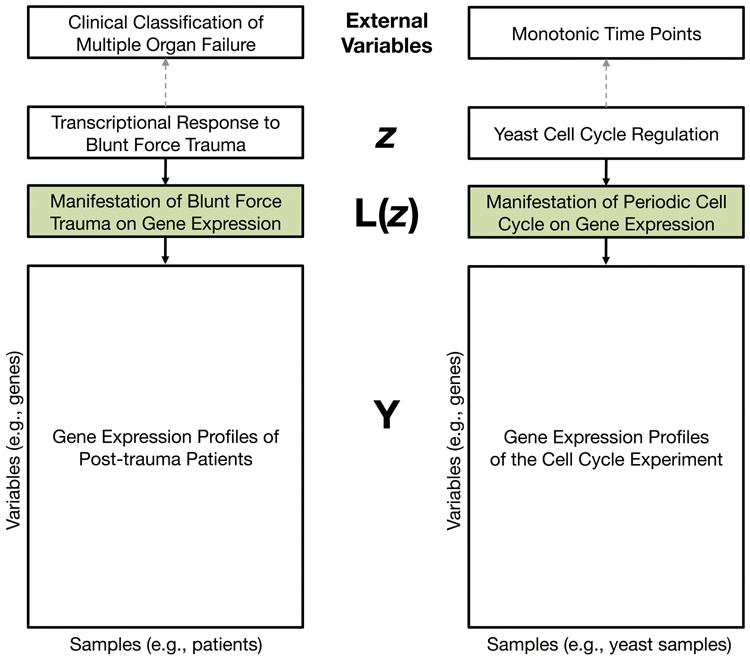
There are a number of well-established methods such as principal component analysis (PCA) for automatically capturing systematic variation due to latent variables in large-scale genomic data. PCA and related methods may directly provide a quantitative characterization of a complex biological variable that is otherwise difficult to precisely define or model. An unsolved problem in this context is how to systematically identify the genomic variables that are drivers of systematic variation captured by PCA. Principal components (PCs) (and other estimates of systematic variation) are directly constructed from the genomic variables themselves, making measures of statistical significance artificially inflated when using conventional methods due to over-fitting.
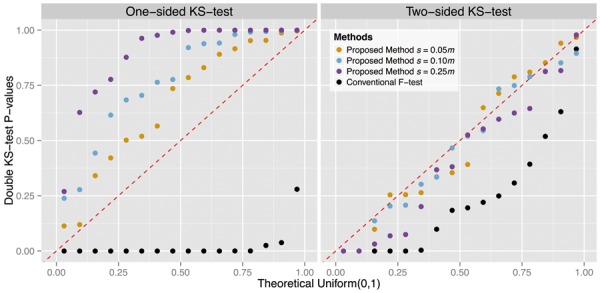
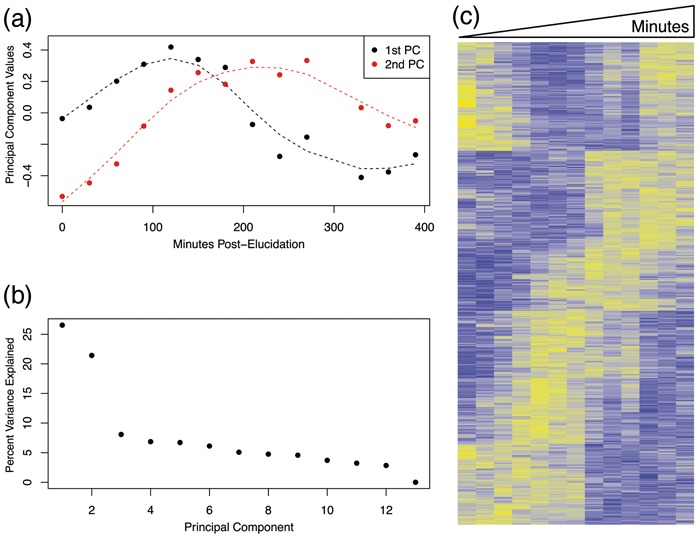
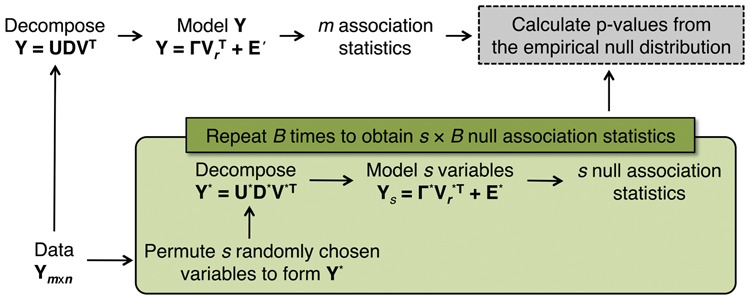
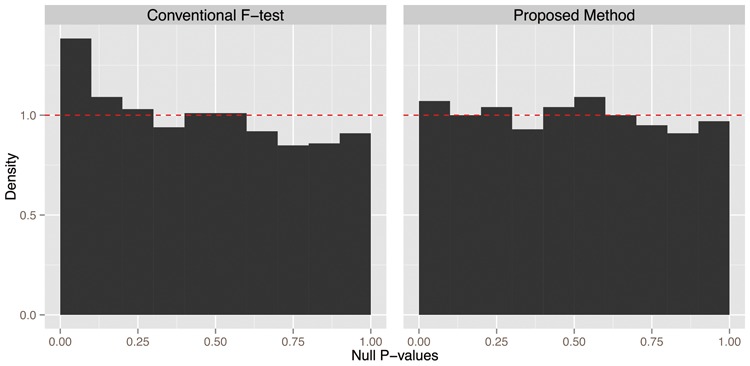
We introduce a new approach called the jackstraw that allows one to accurately identify genomic variables that are statistically significantly associated with any subset or linear combination of PCs. The proposed method can greatly simplify complex significance testing problems encountered in genomics and can be used to identify the genomic variables significantly associated with latent variables. Using simulation, we demonstrate that our method attains accurate measures of statistical significance over a range of relevant scenarios. We consider yeast cell-cycle gene expression data, and show that the proposed method can be used to straightforwardly identify genes that are cell-cycle regulated with an accurate measure of statistical significance. We also analyze gene expression data from post-trauma patients, allowing the gene expression data to provide a molecularly driven phenotype. Using our method, we find a greater enrichment for inflammatory-related gene sets compared to the original analysis that uses a clinically defined, although likely imprecise, phenotype. The proposed method provides a useful bridge between large-scale quantifications of systematic variation and gene-level significance analyses.
An R software package, called jackstraw, is available in CRAN.
有许多成熟的方法,如主成分分析(PCA),用于自动捕捉大规模基因组数据中潜在变量引起的系统变异。PCA及相关方法可以直接对一个复杂的生物学变量进行定量表征,而这个变量用其他方式很难精确界定或建模。在这种情况下,一个尚未解决的问题是如何系统地识别那些驱动PCA所捕捉到的系统变异的基因组变量。主成分(PC)(以及其他系统变异估计值)直接由基因组变量本身构建而成,这使得在使用传统方法时,由于过度拟合,统计显著性的测量值会被人为夸大。
我们引入了一种名为jackstraw的新方法,它能让人们准确识别与PC的任何子集或线性组合在统计上显著相关的基因组变量。所提出的方法可以极大地简化基因组学中遇到的复杂显著性检验问题,并可用于识别与潜在变量显著相关的基因组变量。通过模拟,我们证明了我们的方法在一系列相关场景中都能获得准确的统计显著性测量值。我们考虑了酵母细胞周期基因表达数据,并表明所提出的方法可用于直接识别受细胞周期调控的基因,并能准确测量其统计显著性。我们还分析了创伤后患者的基因表达数据,使基因表达数据提供一种分子驱动的表型。使用我们的方法,与使用临床定义(尽管可能不准确)的表型的原始分析相比,我们发现炎症相关基因集的富集程度更高。所提出的方法在系统变异的大规模量化和基因水平的显著性分析之间架起了一座有用的桥梁。
一个名为jackstraw的R软件包可在CRAN上获取。