Skwark Marcin J, Raimondi Daniele, Michel Mirco, Elofsson Arne
Department of Biochemistry and Biophysics, Stockholm University, Stockholm, Sweden; Science for Life Laboratory, Stockholm University, Solna, Sweden; Department of Information and Computer Science, Aalto University, Aalto, Finland.
Department of Biochemistry and Biophysics, Stockholm University, Stockholm, Sweden; Science for Life Laboratory, Stockholm University, Solna, Sweden; Interuniversity Institute of Bioinformatics in Brussels, ULB-VUB, La Plaine Campus, Triomflaan, Brussels, Belgium.
PLoS Comput Biol. 2014 Nov 6;10(11):e1003889. doi: 10.1371/journal.pcbi.1003889. eCollection 2014 Nov.
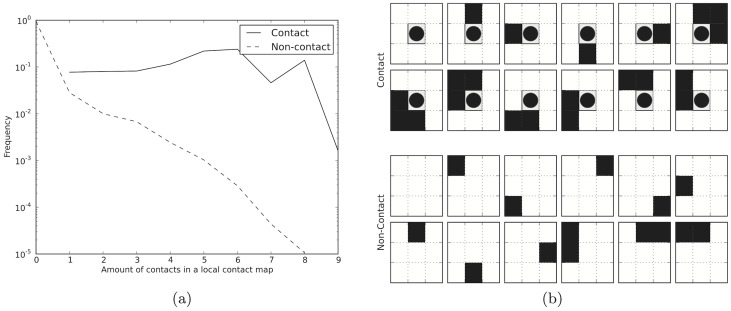
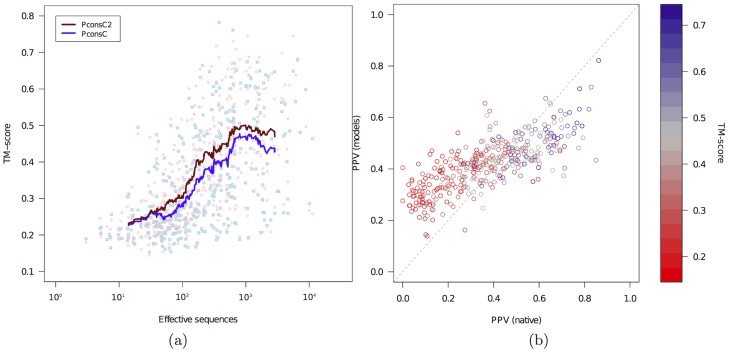
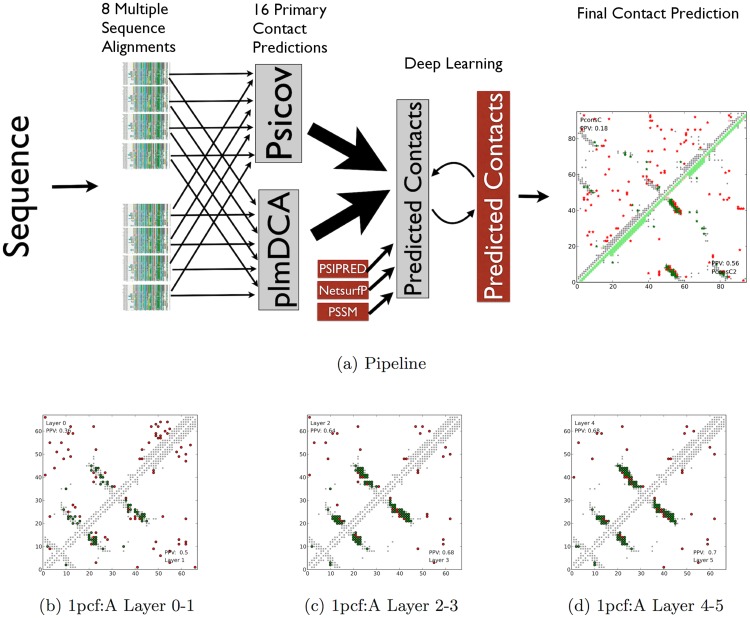
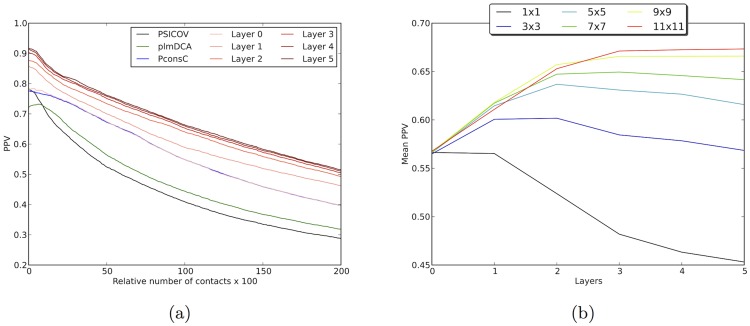
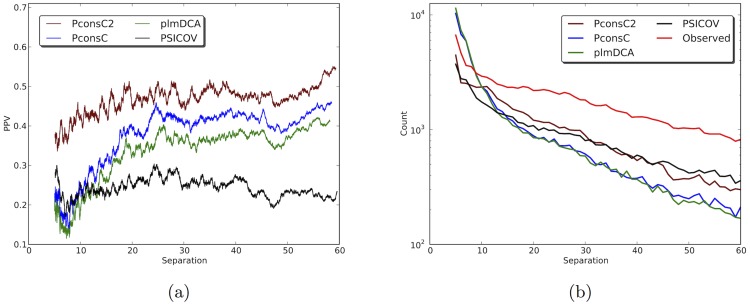
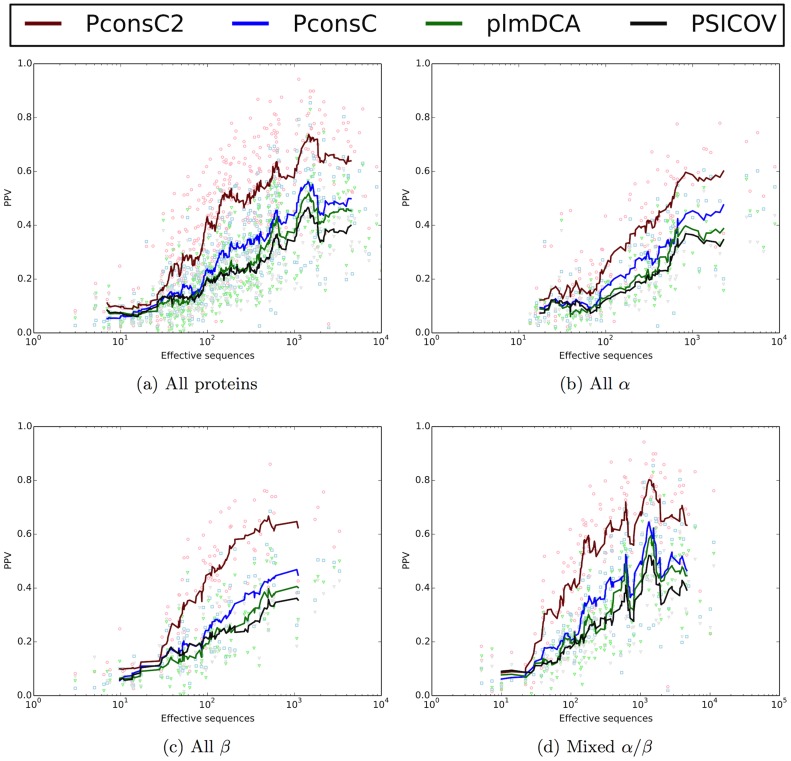
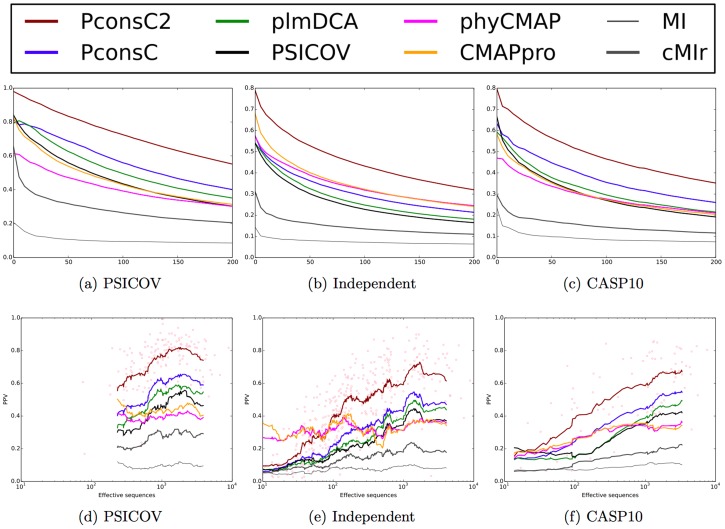
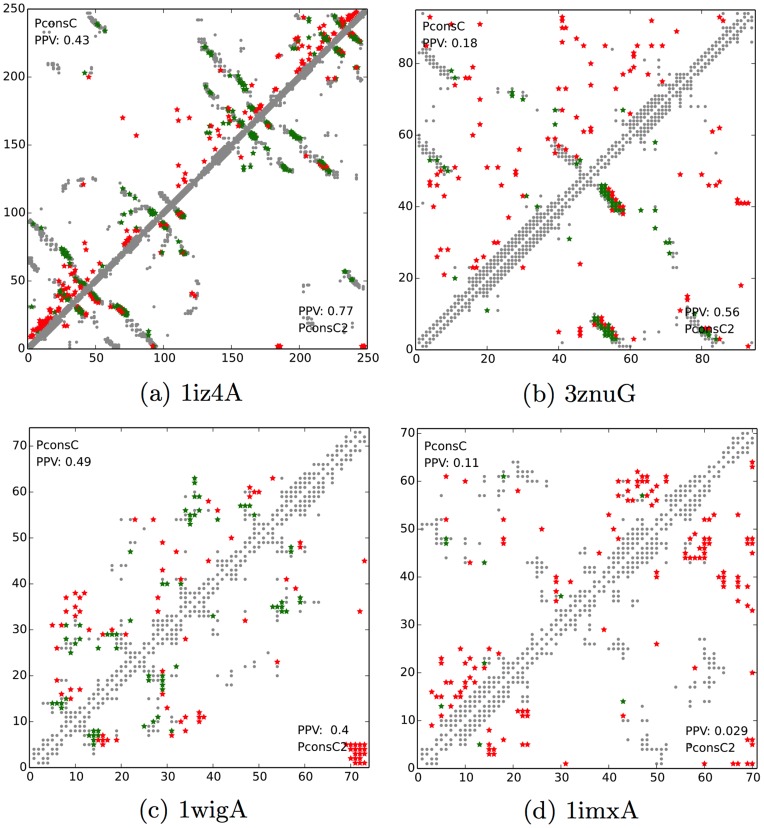
Given sufficient large protein families, and using a global statistical inference approach, it is possible to obtain sufficient accuracy in protein residue contact predictions to predict the structure of many proteins. However, these approaches do not consider the fact that the contacts in a protein are neither randomly, nor independently distributed, but actually follow precise rules governed by the structure of the protein and thus are interdependent. Here, we present PconsC2, a novel method that uses a deep learning approach to identify protein-like contact patterns to improve contact predictions. A substantial enhancement can be seen for all contacts independently on the number of aligned sequences, residue separation or secondary structure type, but is largest for β-sheet containing proteins. In addition to being superior to earlier methods based on statistical inferences, in comparison to state of the art methods using machine learning, PconsC2 is superior for families with more than 100 effective sequence homologs. The improved contact prediction enables improved structure prediction.
给定足够多的大蛋白质家族,并使用全局统计推断方法,在蛋白质残基接触预测中有可能获得足够的准确性,从而预测许多蛋白质的结构。然而,这些方法没有考虑到蛋白质中的接触既不是随机分布,也不是独立分布的,而是实际上遵循由蛋白质结构所支配的精确规则,因此是相互依赖的。在此,我们提出了PconsC2,这是一种使用深度学习方法来识别类似蛋白质的接触模式以改进接触预测的新方法。对于所有接触,无论比对序列的数量、残基间距或二级结构类型如何,都能看到显著的增强,而对于含β折叠的蛋白质增强最大。除了优于基于统计推断的早期方法外,与使用机器学习的现有方法相比,PconsC2对于具有100多个有效序列同源物的家族更具优势。改进的接触预测能够实现改进的结构预测。