Song Li, Florea Liliana, Langmead Ben
Genome Biol. 2014;15(11):509. doi: 10.1186/s13059-014-0509-9.
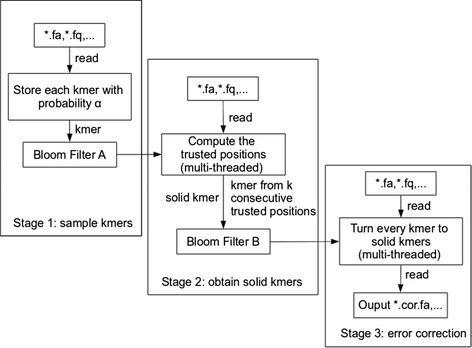
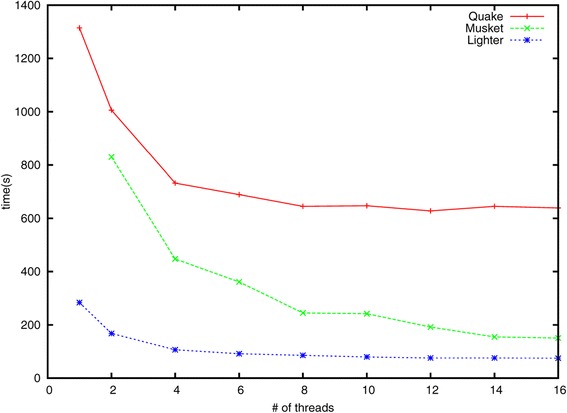
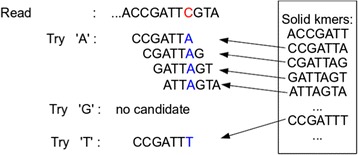
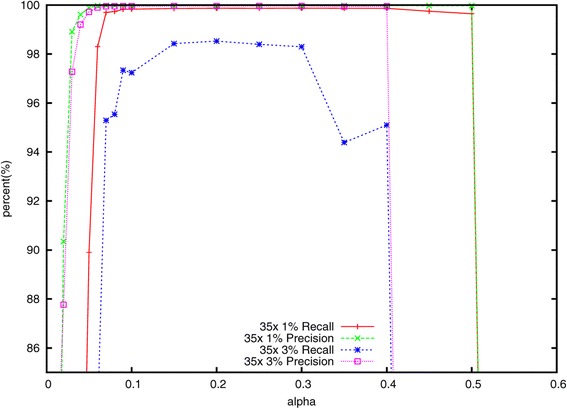
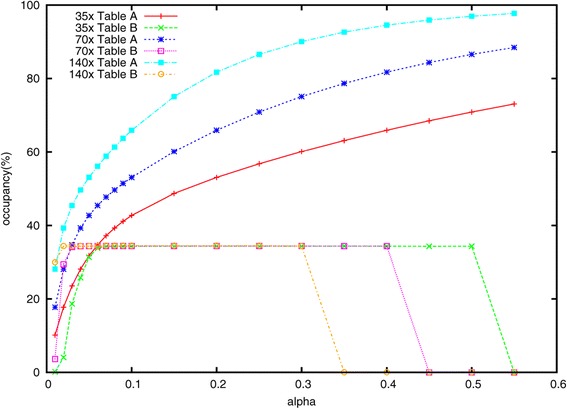
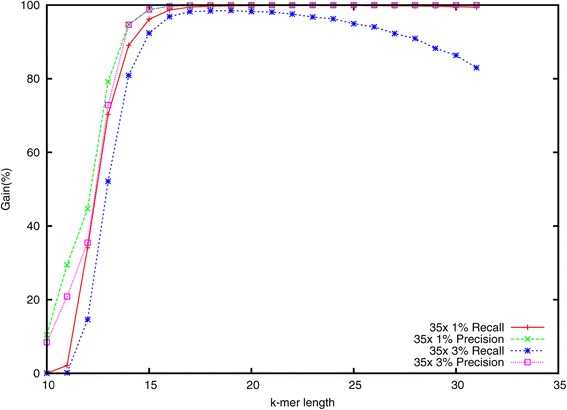
Lighter is a fast, memory-efficient tool for correcting sequencing errors. Lighter avoids counting k-mers. Instead, it uses a pair of Bloom filters, one holding a sample of the input k-mers and the other holding k-mers likely to be correct. As long as the sampling fraction is adjusted in inverse proportion to the depth of sequencing, Bloom filter size can be held constant while maintaining near-constant accuracy. Lighter is parallelized, uses no secondary storage, and is both faster and more memory-efficient than competing approaches while achieving comparable accuracy.
Lighter是一种快速、内存高效的用于纠正测序错误的工具。Lighter避免对k-mer进行计数。相反,它使用一对布隆过滤器,一个保存输入k-mer的样本,另一个保存可能正确的k-mer。只要采样率与测序深度成反比进行调整,布隆过滤器的大小就可以保持不变,同时保持近乎恒定的准确性。Lighter进行了并行化处理,不使用二级存储,并且在实现可比准确性的同时,比其他竞争方法更快且内存效率更高。