Dettman Jeremy R, Rodrigue Nicolas, Kassen Rees
Department of Biology and Centre for Advanced Research in Environmental Genomics, University of Ottawa, Ottawa, Ontario, Canada
Department of Biology, Carleton University, Ottawa, Ontario, Canada.
Genome Biol Evol. 2014 Dec 4;7(1):18-34. doi: 10.1093/gbe/evu260.
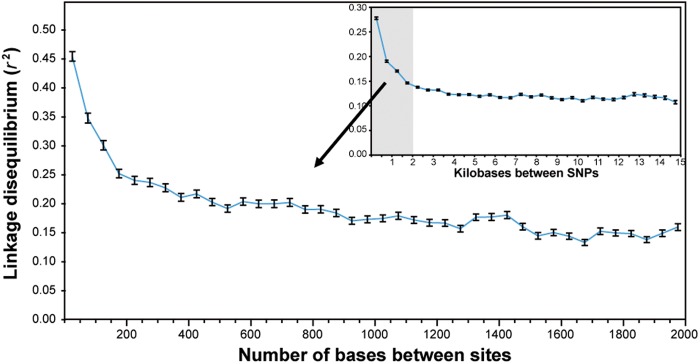
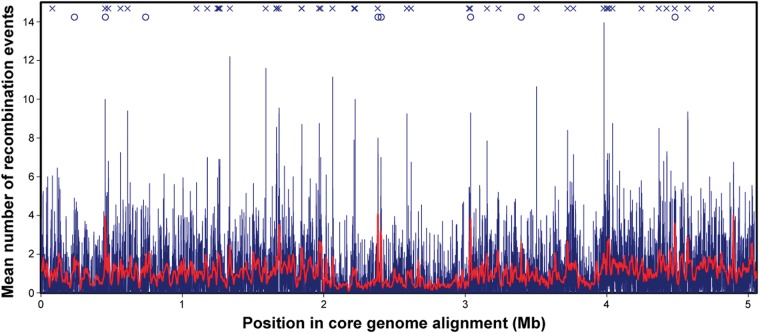
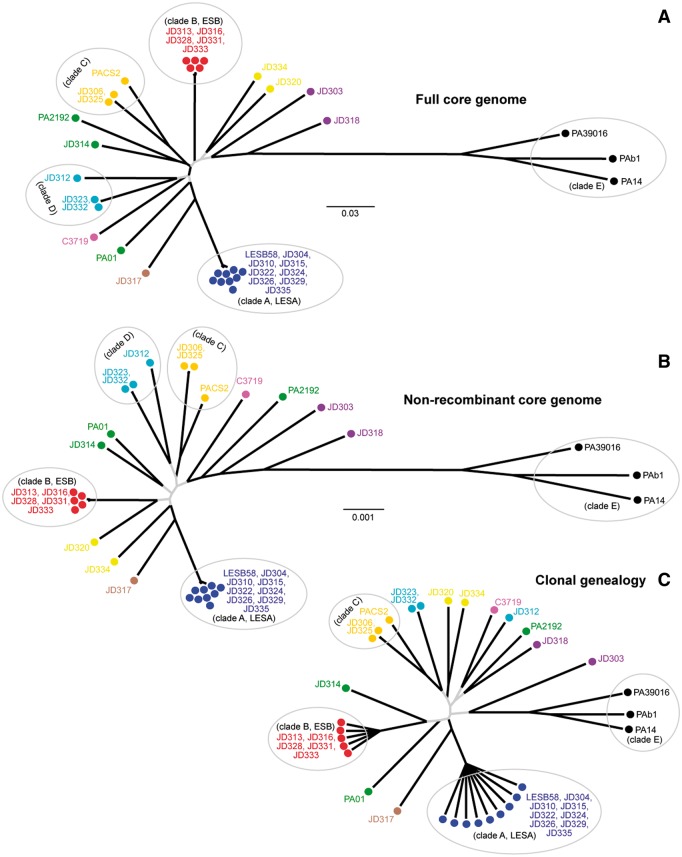
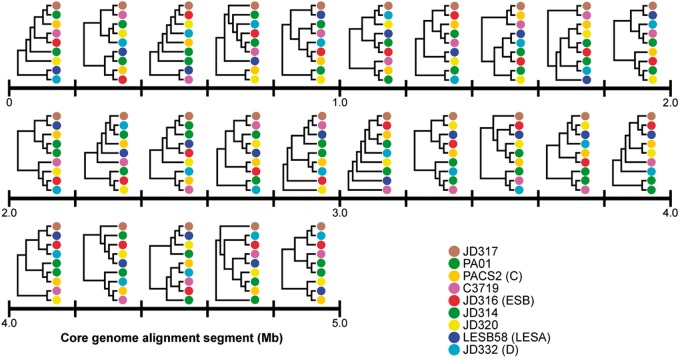
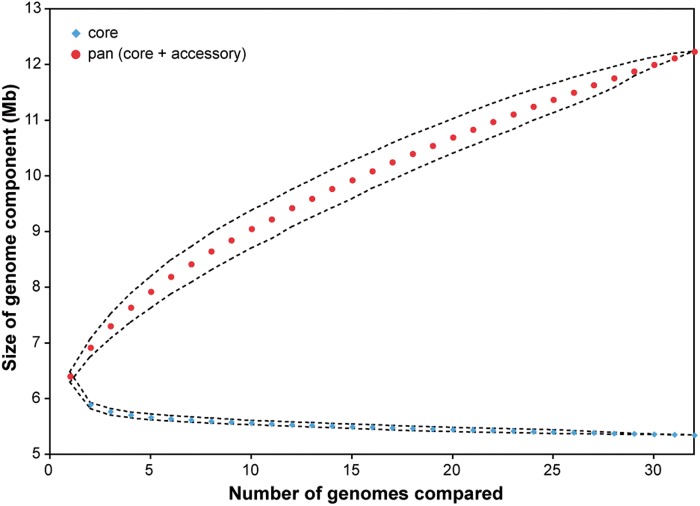
The bacterium Pseudomonas aeruginosa is a significant cause of acute nosocomial infections as well as chronic respiratory infections in patients with cystic fibrosis (CF). Recent reports of the intercontinental spread of a CF-specific epidemic strain, combined with high intrinsic levels of antibiotic resistance, have made this opportunistic pathogen an important public health concern. Strain-specific differences correlate with variation in clinical outcomes of infected CF patients, increasing the urgency to understand the evolutionary origin of genetic factors conferring important phenotypes that enable infection, virulence, or resistance. Here, we describe the genome-wide patterns of homologous and nonhomologous recombination in P. aeruginosa, and the extent to which the genomes are affected by these diversity-generating processes. Based on whole-genome sequence data from 32 clinical isolates of P. aeruginosa, we examined the rate and distribution of recombination along the genome, and its effect on the reconstruction of phylogenetic relationships. Multiple lines of evidence suggested that recombination was common and usually involves short stretches of DNA (200-300 bp). Although mutation was the main source of nucleotide diversity, the import of polymorphisms by homologous recombination contributed nearly as much. We also identified the genomic regions with frequent recombination, and the specific sequences of recombinant origin within epidemic strains. The functional characteristics of the genes contained therein were examined for potential associations with a pathogenic lifestyle or adaptation to the CF lung environment. A common link between many of the high-recombination genes was their functional affiliation with the cell wall, suggesting that the products of recombination may be maintained by selection for variation in cell-surface molecules that allows for evasion of the host immune system.
铜绿假单胞菌是急性医院感染以及囊性纤维化(CF)患者慢性呼吸道感染的重要病因。最近有报道称一种CF特异性流行菌株在洲际传播,再加上其固有的高抗生素耐药水平,使得这种机会致病菌成为一个重要的公共卫生问题。菌株特异性差异与感染CF患者的临床结果差异相关,这增加了了解赋予感染、毒力或耐药等重要表型的遗传因素进化起源的紧迫性。在此,我们描述了铜绿假单胞菌全基因组范围内同源和非同源重组的模式,以及基因组受这些产生多样性过程影响的程度。基于32株铜绿假单胞菌临床分离株的全基因组序列数据,我们研究了重组在基因组中的速率和分布,及其对系统发育关系重建的影响。多条证据表明重组很常见,通常涉及短片段DNA(200 - 300 bp)。虽然突变是核苷酸多样性的主要来源,但同源重组导入的多态性贡献也几乎相同。我们还确定了频繁重组的基因组区域,以及流行菌株中重组起源的特定序列。对其中所含基因的功能特征进行了检查,以寻找与致病生活方式或对CF肺部环境适应性的潜在关联。许多高重组基因之间的一个共同联系是它们在功能上与细胞壁相关,这表明重组产物可能通过选择细胞表面分子的变异得以维持,从而实现逃避免疫系统。