Li Shijun, Sun Qiangzheng, Wei Xiaoyu, Klena John D, Wang Jianping, Liu Ying, Tian Kecheng, Luo Xia, Ye Changyun, Xu Jianguo, Wang Dingming, Tang Guangpeng
Institute of Communicable Disease Control and Prevention, Guizhou Provincial Center for Disease Control and Prevention, 101 Bageyan Road, Guiyang, 550004, Guizhou, People's Republic of China.
State Key Laboratory for Infectious Disease Prevention and Control, Collaborative Innovation Center for Diagnosis and Treatment of Infectious Diseases, National Institute for Communicable Disease Control and Prevention, China CDC, P.O. Box 5, Changping, Beijing, China.
PLoS One. 2015 Jan 24;10(1):e0116708. doi: 10.1371/journal.pone.0116708. eCollection 2015.
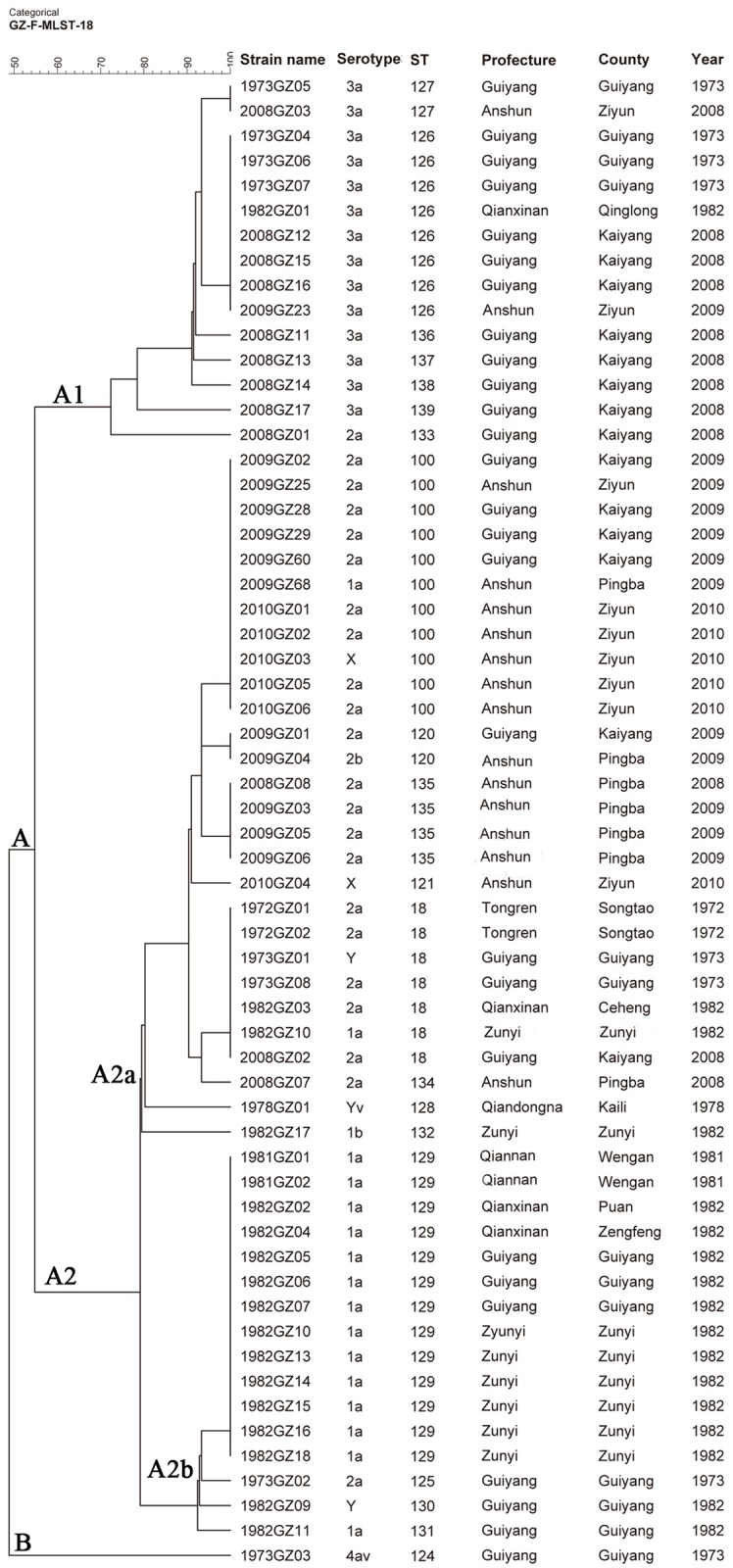
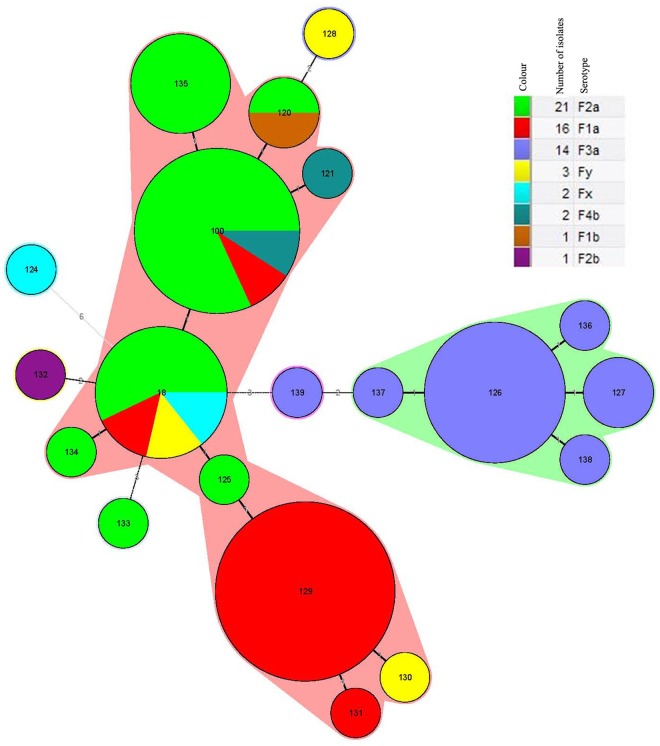
Shigella flexneri is one of the major etiologic causes of shigellosis in Guizhou Province, China. However, the genetic characteristics of circulating isolates are unknown. Phenotypic and molecular profiles of 60 S. flexneri isolates recovered in Guizhou between 1972 to 1982 and 2008 to 2010 were determined. Nine serotypes (1a, 2a, 3a, 1b, 2b, X, Y, 4av and Yv) were identified. Multi-locus sequence typing differentiated the isolates into 20 sequence types (STs); 18 were novel. Four STs, ST 129, ST 100, ST 126 and ST 18, were most abundant, accounting for 65% of the isolates. Thirty-nine NotI-pulsed field gel electrophoresis patterns (pulsotypes, PTs) were observed; eight PTs were represented by more than one isolate with six isolates sharing the PT 13 profile. Multi-locus variable-nucleotide tandem-repeat analysis recognized 44 different types (MTs); seven MTs were represented by more than one isolate and MT 1 was most commonly encountered. Correlation between genetic relationships and serotypes was observed among the isolates studied; the majority of isolates belonging to the same serotype from different years clustered together based on the molecular data. These clustered isolates were also from similar geographical origins. These results enhance our understanding of genetic relationships between S. flexneri in Guizhou Province and can be used to help understand the changing etiology of shigellosis in China.
福氏志贺菌是中国贵州省志贺菌病的主要病因之一。然而,流行菌株的遗传特征尚不清楚。我们测定了1972年至1982年以及2008年至2010年期间在贵州分离出的60株福氏志贺菌的表型和分子特征。鉴定出9种血清型(1a、2a、3a、1b、2b、X、Y、4av和Yv)。多位点序列分型将这些分离株分为20种序列类型(STs);其中18种是新的。四种STs,即ST 129、ST 100、ST 126和ST 18最为常见,占分离株的65%。观察到39种NotI脉冲场凝胶电泳图谱(脉冲型,PTs);8种PTs由不止一株分离株代表,6株分离株具有相同的PT 13图谱。多位点可变核苷酸串联重复分析识别出44种不同类型(MTs);7种MTs由不止一株分离株代表,MT 1最为常见。在所研究的分离株中观察到遗传关系与血清型之间的相关性;根据分子数据,来自不同年份的大多数属于同一血清型的分离株聚集在一起。这些聚集的分离株也来自相似的地理区域。这些结果增进了我们对贵州省福氏志贺菌遗传关系的理解,并可用于帮助了解中国志贺菌病病因的变化。