Krieger Elmar, Vriend Gert
Centre for Molecular and Biomolecular Informatics, Radboudumc, PO Box 9101, 6500 HB Nijmegen, The Netherlands.
J Comput Chem. 2015 May 15;36(13):996-1007. doi: 10.1002/jcc.23899. Epub 2015 Mar 30.
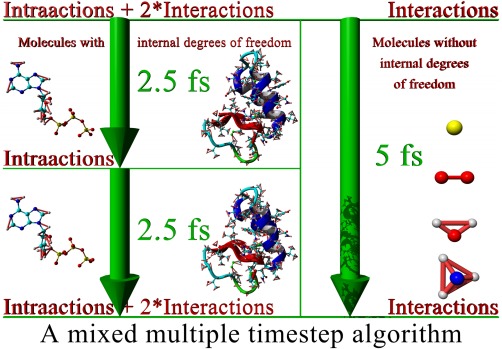
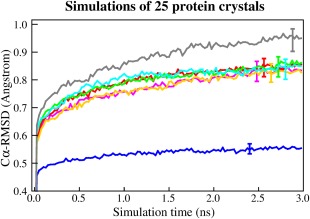
We describe a set of algorithms that allow to simulate dihydrofolate reductase (DHFR, a common benchmark) with the AMBER all-atom force field at 160 nanoseconds/day on a single Intel Core i7 5960X CPU (no graphics processing unit (GPU), 23,786 atoms, particle mesh Ewald (PME), 8.0 Å cutoff, correct atom masses, reproducible trajectory, CPU with 3.6 GHz, no turbo boost, 8 AVX registers). The new features include a mixed multiple time-step algorithm (reaching 5 fs), a tuned version of LINCS to constrain bond angles, the fusion of pair list creation and force calculation, pressure coupling with a "densostat," and exploitation of new CPU instruction sets like AVX2. The impact of Intel's new transactional memory, atomic instructions, and sloppy pair lists is also analyzed. The algorithms map well to GPUs and can automatically handle most Protein Data Bank (PDB) files including ligands. An implementation is available as part of the YASARA molecular modeling and simulation program from www.YASARA.org.
我们描述了一组算法,这些算法能够在单个英特尔酷睿i7 5960X CPU(无图形处理单元(GPU),23,786个原子,粒子网格埃瓦尔德(PME),8.0 Å截止值,正确的原子质量,可重现轨迹,3.6 GHz的CPU,无睿频加速,8个AVX寄存器)上,使用AMBER全原子力场,以每天160纳秒的速度模拟二氢叶酸还原酶(DHFR,一个常见的基准)。新特性包括一种混合多时间步算法(可达5飞秒)、经过调整的LINCS以约束键角、对列表创建与力计算的融合、使用“密度调节器”进行压力耦合以及利用AVX2等新的CPU指令集。还分析了英特尔新的事务性内存、原子指令和宽松对列表的影响。这些算法能很好地映射到GPU上,并且可以自动处理大多数包括配体的蛋白质数据库(PDB)文件。作为来自www.YASARA.org的YASARA分子建模与模拟程序的一部分,有该算法的实现版本可供使用。