Xue Li C, Jordan Rafael A, El-Manzalawy Yasser, Dobbs Drena, Honavar Vasant
Bioinformatics and Computational Biology Program, Iowa State University, Ames, IA, 50011, USA.
Department of Genetics, Development and Cell Biology, Iowa State University, Ames, 50011, USA.
ACM BCB. 2011 Aug;2011:441-445. doi: 10.1145/2147805.2147866.
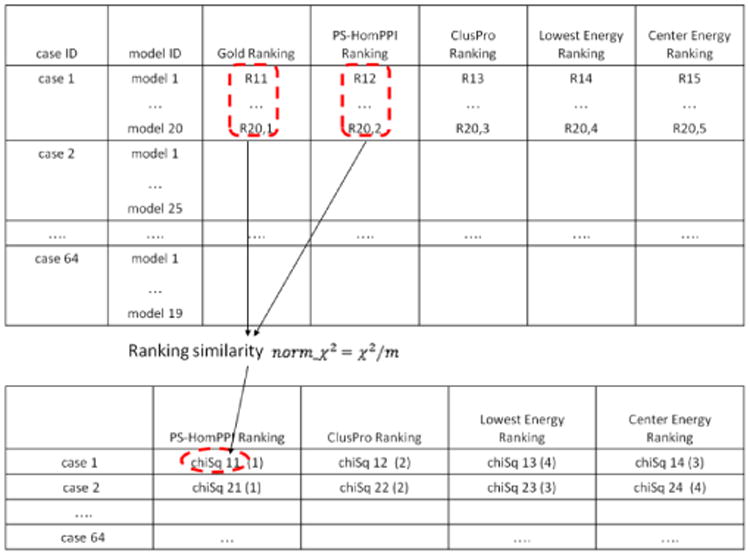
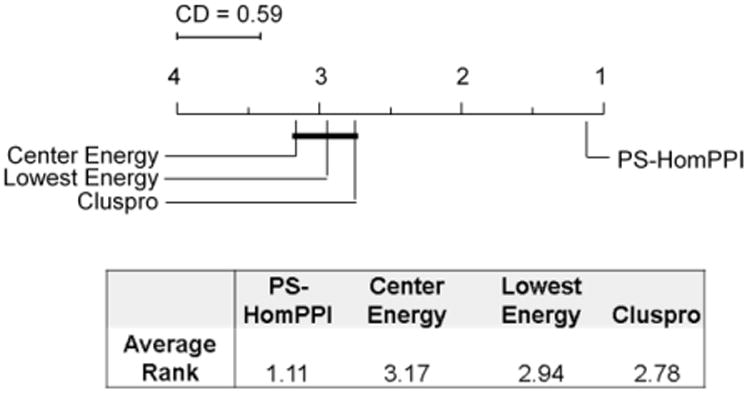
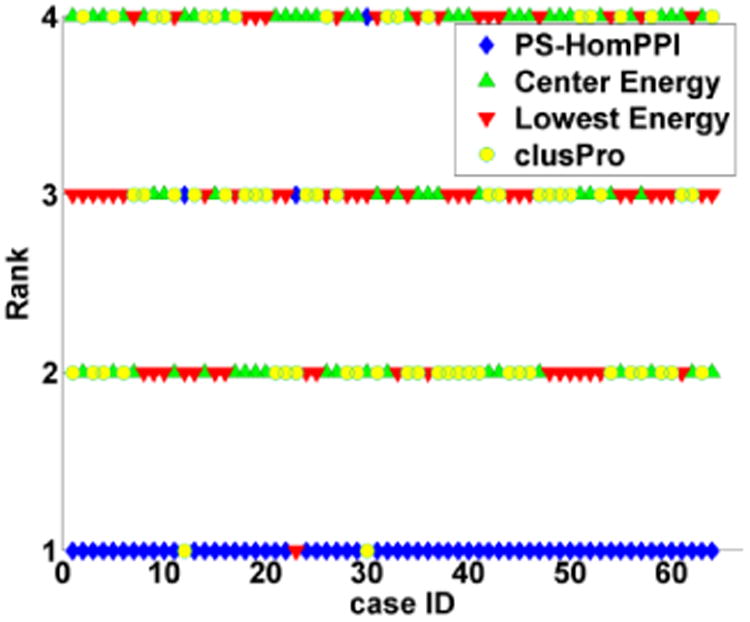
Computational protein-protein docking is a valuable tool for determining the conformation of complexes formed by interacting proteins. Selecting near-native conformations from the large number of possible models generated by docking software presents a significant challenge in practice. We introduce a novel method for ranking docked conformations based on the degree of overlap between the interface residues of a docked conformation formed by a pair of proteins with the set of predicted interface residues between them. Our approach relies on a method, called PS-HomPPI, for reliably predicting protein-protein interface residues by taking into account information derived from both interacting proteins. PS-HomPPI infers the residues of a query protein that are likely to interact with a partner protein based on known interface residues of the homo-interologs of the query-partner protein pair, i.e., pairs of interacting proteins that are homologous to the query protein and partner protein. Our results on Docking Benchmark 3.0 show that the quality of the ranking of docked conformations using our method is consistently superior to that produced using ClusPro cluster-size-based and energy-based criteria for 61 out of the 64 docking complexes for which PS-HomPPI produces interface predictions. An implementation of our method for ranking docked models is freely available at: http://einstein.cs.iastate.edu/DockRank/.
计算蛋白质-蛋白质对接是确定相互作用蛋白质形成的复合物构象的一种有价值的工具。在实践中,从对接软件生成的大量可能模型中选择接近天然的构象是一项重大挑战。我们引入了一种基于一对蛋白质形成的对接构象的界面残基与它们之间预测的界面残基集之间的重叠程度来对接合构象进行排名的新方法。我们的方法依赖于一种称为PS-HomPPI的方法,该方法通过考虑来自两个相互作用蛋白质的信息来可靠地预测蛋白质-蛋白质界面残基。PS-HomPPI根据查询-伙伴蛋白质对的同系物相互作用体(即与查询蛋白质和伙伴蛋白质同源的相互作用蛋白质对)的已知界面残基,推断查询蛋白质中可能与伙伴蛋白质相互作用的残基。我们在对接基准3.0上的结果表明,对于PS-HomPPI产生界面预测的64个对接复合物中的61个,使用我们的方法对接合构象进行排名的质量始终优于使用基于ClusPro聚类大小和能量的标准所产生的质量。我们对接合模型进行排名的方法的实现可在以下网址免费获取:http://einstein.cs.iastate.edu/DockRank/ 。